Phaser–1.2
Phaser is crystallographic software for phasing macromolecular crystal structures
with maximum likelihood techniques. It is available through the Phenix software
suite, and directly from the authors. It will also be in future releases of the
CCP4 software suite.
Index to Documentation
Version History
- Phaser–1.2
- First "official" release of Phaser software
- Automated solution of structures
- New Likelihood Enhanced fast Translation Function (LETF)
- Ability to use electron density maps as molecular replacement models
- Rigid body solution refinement against maximum likelihood target
- Pruning of duplicate solutions from solution list
- Searching of multiple alternative space groups
- Searching of multiple alternative models
- Better minimizers
- Phaser–1.1
- Bug in anisotropy refinement corrected
- Phaser–1.0
1. Introduction
This is the documentation for running Phaser–1.2 with keyword input. Please
note that keyword input has changed considerably since Phaser–1.1. Your old
scripts will not work with Phaser–1.2!
1.1 Keyword Index
BINS BOXScale
CELL CLMN COMPosition
ENSEmble FINAl HKLIn
HKLOut IPLN LABIn
MODE MRMAcrocycle OUTLier
NORMalisation PACK PERMutations
RESCore RESOlution ROOT
ROTAte SAMPling SAVE
SCRIpt SEARch SELEct
SGALternative SHANnon SNMAcrocycle
SOLUtion SPACegroup SUITe
TARGet TITLe TOPFiles
TRANslate VERBose XYZOut
1.2 Syntax of Documentation
KEYWord
Ordinary type in blue means a keyword. Only the first four letters of any
input keyword (occasionally only three) are required/recognized. The required
characters are given in uppercase and those not required in lowercase, but
keywords are not case sensitive.
<PARAMETER>
Angle brackets mean a parameter value. Input strings are case sensitive:
the case of titles and filenames is preserved.
{ KEYWord
<X Y Z> }
Curly brackets mean a group of keywords/parameters must come together.
[ X | Y ]
Square brackets and line separating options means X or Y.
KEYWord
<X Y Z>
Italics mean the keywords/input is optional.
1.3 Bug Reports
We apologise for the bugs. Please send bug reports to
cimr-phaser@lists.cam.ac.uk
or rjr27@cam.ac.uk
(Randy Read).
2. Molecular Replacement
Phaser should be able to solve most structures with the Automatic
mode, and this is the first mode that you should try. Give Phaser your data (How
to Define Data) and your models (How to Define Models),
and tell Phaser what to search for (use SEARch
keyword). If this doesn't work, you can try selecting peaks of lower significance
in the rotation function in case the real orientation was not within the selection
criteria (by default peaks above 75% of the top peak are selected - use FINAl
keyword to change this).
If the Automatic mode doesn't work you need to run the modes
of phaser separately. The possibilities are endless - you can try brute searches
with the full likelihood target, finer searches, exhaustive searches (translations
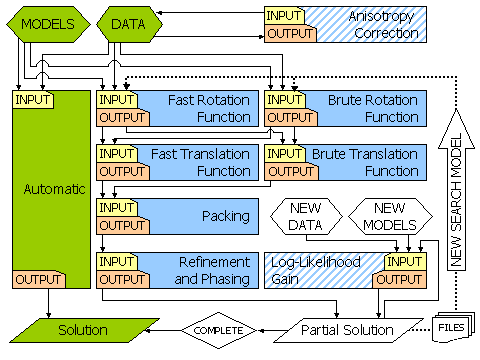
of all orientations) etc. etc. The flow diagram below shows a basic set of protocols
for obtaining a molecular replacement solution with Phaser. The path for Automatic
is shown in green. Other possible paths (with other modes) are shown in blue.
The hatching indicates nonessential modes. The dotted lines show the loop that
allows a full molecular replacement solution to be built up when there is more
that one model in the asymmetric unit.
2.1 How to Define Data
You need to tell Phaser the name of the mtz file containing your data and the
columns in the mtz file to be used.
Additional keywords define how the data are used
OPTIONAL Keywords for Data
BINS
{MINimum <L>} {MAXimum
<H>} {NUMber <N>} {WIDTh
<W>} {CUBIc <A B C>}
CELL <A B C ALPHA BETA GAMMA>
OUTLier [{ON <OUTLIER_PROB>} | OFF]
RESOlution <HIRES> <LORES>
SELEction [ALL | CENTric
| ACENtric | {RANDom
<PC>}]
SPACegroup <SG>
2.2 How To Define Models
Molecular replacement models are defined with the ENSEmble
keyword and the COMPosition
keyword. To compute a Sigma(A) curve representing the accuracy of model structure
factors as a function of resolution, Phaser needs to know the RMS coordinate
error expected for the model (determined directly from RMS or indirectly from
IDENt in the ENSEmble keyword) and the fraction of the scattering power in the
asymmetric unit that this model contributes (deduced from the COMPosition keywords).
If fp is the fraction scattering and RMS is the rms coordinate error, then
Sigma(A) = SQRT{fp*[1-fsol*exp(-Bsol*(sin(theta)/lambda)2)]}
* exp{-(8 Pi2/3)*RMS2*(sin(theta)/lambda)2}
where fsol(=0.95) and Bsol(=300Å2) account for the effects
of disordered solvent.
2.2.1 Ensembles
Phaser must be given the models that it will use for molecular replacement. A
molecular replacement model is constructed in one of two ways - either by making
an Ensemble from a set of aligned homologous structures, entered as pdb files,
or by entering a model from a map, entered as structure factors in an mtz file.
Each ensemble is treated as a separate type of rigid body to be placed in the
molecular replacement solution. An ensemble should only be defined once, even
if there are several copies of the molecule in the asymmetric unit.
Examples of Coordinates as a Model
- You have one structure as a model with 44% sequence identity to the protein
in the crystal.
- ENSEmble
mol1 PDB homology1.pdb IDENtity
.44
- You have three structures as models with 44%, 39% and 35% identity to
the protein in the crystal.
- ENSEmble
mol2 PDB
homology1.pdb IDENtity .44 PDB
homology2.pdb IDENtity .39 PDB
homology3.pdb IDENtity .35
- You have an NMR Ensemble as a model. There is no need to split the coordinates
in the pdb file provided that the models are separated by MODEL and ENDMDL
cards. In this case the homology is not a good indication of the similarity
of the structural coordinates to the target structure. You should use the
RMS option and use an RMS value of at least 1.5Å.
- ENSEmble
mol3 PDB nmr.pdb RMS
1.5
Examples of a Map as a Model
- You have low resolution electron density of your model. This density
has been cut out and converted to structure factors in a large cell.
- ENSEmble
mol1 HKLIn mol1.mtz F
= Fmol1 P = Pmol1 PROTein
MW 10241 NUCLeic_acid MW 0 EXTEnt
23 25 29 RMS 2.0 CENTre
4 3 30
2.2.2 Composition
Phaser must know what percentage of the scattering is given by each Ensemble.
It can not work this out without knowing the content of the asymmetric unit. The
composition of the asymmetric unit is defined in one of two ways - either by entering
the molecular weight in the asymmetric unit, or by entering the fraction scattering
for each Ensemble directly. If entering the composition by molecular weights,
you have to define the molecular weight(s) you give as either protein or nucleic
acid, since protein and nucleic acids scatter differently. In this case, the total
composition is the sum of all the compositions given. If entering the composition
as a fraction of the scattering, the total fraction scattering must be less than
one.
Examples of Composition by Molecular Weight
- You have one protein with MW 21022 in the asymmetric unit
- COMPosition PROTein
MW 21022
- You have three copies of a protein with MW 21022 in the asymmetric unit
- COMPosition PROTein
MW 21022
- COMPosition PROTein
MW 21022
- COMPosition PROTein
MW 21022
- Another way of entering the same thing is
- COMPosition PROTein
MW 21022 NUMber
3
- Yet another way of entering the same thing is
- COMPosition PROTein
MW 63066
- You have two copies of a protein with MW 21022, two copies of a protein
with MW 9843 and RNA with MW 32004 in the asymmetric unit
- COMPosition PROTein
MW 21022 NUMber
2
- COMPosition PROTein
MW 9843 NUMber
2
- COMPosition NUCLeic_acid
MW 32004
Examples of Composition by Percentage Scattering
- Each copy of Ensemble mol1 gives 22% of the scattering
- COMPosition ENSEmble
mol1 FRACtional 0.22
- Each copy of Ensemble mol2 gives 78% of the scattering
- COMPosition ENSEmble
mol2 FRACtional 0.78
Additional parameters define the details of how the ensembling is done and the
completeness is modeled. See documentation for the individual keywords for more
information.
OPTIONAL Keywords for Models
2.3 How To Define Solutions
You don't really need to know how to define molecular replacement solutions as
Phaser writes out files ending in .sol
and .rlist that contain the solution
information from the job. The root of the files is given by the ROOT
keyword. By default, the root filename is PHASER. These files can be read back
into subsequent runs of Phaser to build up solutions containing more than one
molecule in the asymmetric unit.
"PHASER.sol" files
are generated by all modes, and contain the current idea of potential molecular
replacement solutions.
"PHASER.rlist" files
are generated by the rotation function modes, and are for performing translation
functions. (They are also produced by degenerate (2D) translation functions,
for performing a translation function to find the third dimension)
To include the files you should use the preprocessor
command @
@
filename.sol
@
filename.rlist
However, if you want to understand "PHASER.sol"
and "PHASER.rlist" files,
read on…
2.3.1 PHASER.sol
At different stages of molecular replacement, an Ensemble will be oriented but
not positioned (after the rotation search), or oriented and positioned (after
the translation search), or, rarely, oriented and the position in 2 of 3 dimensions
known. These three states correspond to solutions defined by the keywords SOLUtion
6DIM, SOLUtion
3DIM, and SOLUtion
5DIM. Each Ensemble in the asymmetric unit has its own SOLUtion
keyword. Examples of the different types of molecular replacement solutions
are:
- One copy of mol1 with known orientation and position (fractional coordinates)
- SOLUtion 6DIM
ENSEmble mol1 EULEr
17 20 32 FRACtional 0.12 0.05 0.74
- One copy of mol1 with known orientation only
- SOLUtion 3DIM
ENSEmble mol1 EULEr
17 20 32
- One copy of mol1 and one copy of mol2 both with known orientation and
position (orthogonal coordinates)
- SOLUtion 6DIM
ENSEmble mol1 EULEr
17 20 32 ORTHogonal 22 18 50
- SOLUtion 6DIM
ENSEmble mol2 EULEr
5 183 230 ORTHogonal 22 11 68
- One copy of mol1 with known orientation and position (fractional coordinates)
and one copy of mol2 with known orientation only
- SOLUtion 6DIM
ENSEmble mol1 EULEr
17 20 32 FRACtional 0.12 0.05 0.74
- SOLUtion 3DIM
ENSEmble mol2 EULEr
5 183 230
- Two copies of mol1 with known orientation and position (fractional coordinates),
one copy of mol2 with known orientation and position (fractional coordinates)
and one copy of mol2 with known orientation only
- SOLUtion 6DIM
ENSEmble mol1 EULEr
17 20 32 FRACtional 0.12 0.05 0.74
- SOLUtion 6DIM
ENSEmble mol1 EULEr
24 23 24 FRACtional 0.58 0.73 0.93
- SOLUtion 3DIM
ENSEmble mol2 EULEr
68 7 85 FRACtional 0.04 0.19 0.25
- SOLUtion 3DIM
ENSEmble mol2 EULEr
5 183 230
When more than one molecular replacement solution is present, the solutions
are separated with the SOLUTION
SET keywords. For example, the 5 types of solution above could be entered
into the same file as
- SOLUtion SET
- SOLUtion 6DIM
ENSEmble mol1 EULEr
17 20 32 FRACtional 0.12 0.05 0.74
- SOLUtion SET
- SOLUtion
3DIM ENSEmble mol1 EULEr
17 20 32
- SOLUtion SET
- SOLUtion 6DIM
ENSEmble mol1 EULEr
17 20 32 ORTHogonal 22 18 50
- SOLUtion 6DIM
ENSEmble mol2 EULEr
5 183 230 ORTHogonal 22 11 68
- SOLUtion SET
- SOLUtion 6DIM
ENSEmble mol1 EULEr
17 20 32 FRACtional 0.12 0.05 0.74
- SOLUtion 3DIM
ENSEmble mol2 EULEr
5 183 230
- SOLUtion SET
- SOLUtion 6DIM
ENSEmble mol1 EULEr
17 20 32 FRACtional 0.12 0.05 0.74
- SOLUtion 6DIM
ENSEmble mol1 EULEr
24 23 24 FRACtional 0.58 0.73 0.93
- SOLUtion 6DIM
ENSEmble mol2 EULEr
68 7 85 FRACtional 0.04 0.19 0.25
- SOLUtion 3DIM
ENSEmble mol2 EULEr
5 183 230
Phaser then performs the functionality of the mode for each solution set.
However, it is much more likely that at any given stage in the structure solution
all the solutions will have the same number of ensembles oriented, and/or oriented
and positioned, and the the solutions will look very similar.. For example,
if the rotation function and translation function for mol1 were very clear,
then there will only be one type of 6DIM
solution for mol1. If the rotation and translation functions for mol2 were then
not clear, there will be a series of possible 6DIM
solutions for mol2.
- SOLUtion SET
- SOLUtion 6DIM
ENSEmble mol1 EULEr
17 20 32 FRACtional 0.12 0.05 0.74
- SOLUtion 6DIM
ENSEmble mol2 EULEr
5 183 230 FRACtional 0.71 0.54 0.81
- SOLUtion SET
- SOLUtion 6DIM
ENSEmble mol1 EULEr
17 20 32 FRACtional 0.12 0.05 0.74
- SOLUtion 6DIM
ENSEmble mol2 EULEr
51 93 75 FRACtional 0.08 0.57 0.25
- SOLUtion SET
- SOLUtion 6DIM
ENSEmble mol1 EULEr
17 20 32 FRACtional 0.12 0.05 0.74
- SOLUtion 3DIM
ENSEmble mol2 EULEr
5 33 21 FRACtional 0.32 0.05 0.44
- SOLUtion SET
- SOLUtion 6DIM
ENSEmble mol1 EULEr
17 20 32 FRACtional 0.12 0.05 0.74
- SOLUtion 3DIM
ENSEmble mol2 EULEr
94 45 91 FRACtional 0.42 0.46 0.55
Where only the coordinates in 2 dimensions (a plane through the origin) of an
oriented Ensemble are determined, a solution of type 5DIM
is produced. The degenerate direction is defined as the direction perpendicular
to the plane in which the position is given. These solutions can be treated
in exactly the same way as the 3DIM
and 6DIM solutions
SOLUtion 5DIM ENSEmble
mol1 EULEr 17 20 32 DEGEnerate
X FRACtional 0.05 0.74
2.3.2 PHASER.rlist
These files define a rotation function list. The peak list is given with a series
of SOLUtion TRIAl
keywords.
SOLUtion TRIAl ENSEmble
mol1 EULEr 17 20 32
SOLUtion TRIAl ENSEmble
mol1 EULEr 67 65 51
SOLUtion TRIAl ENSEmble
mol1 EULEr 67 112 81
SOLUtion TRIAl ENSEmble
mol1 EULEr 68 19 38
If a partial solution is already known, then the information for the currently
"known" parts of the asymmetric unit is given in the form used for
the PHASER.sol file, followed
by the list of trial orientations for which a translation function is to be
performed.
SOLUtion SET
SOLUtion 6DIM ENSEmble
mol1 EULEr 17 20 32 FRACtional
0.12 0.05 0.74
SOLUtion TRIAl ENSEmble
mol1 EULEr 44 20 32
SOLUtion TRIAl ENSEmble
mol1 EULEr 67 65 51
SOLUtion TRIAl ENSEmble
mol1 EULEr 67 112 81
SOLUtion TRIAl ENSEmble
mol1 EULEr 68 19 38
SOLUtion SET
SOLUtion 6DIM ENSEmble
mol1 EULEr 17 20 32 FRACtional
0.13 0.55 0.76
SOLUtion TRIAl ENSEmble
mol1 EULEr 83 9 180
SOLUtion TRIAl ENSEmble
mol1 EULEr 8 36 92
SOLUtion TRIAl ENSEmble
mol1 EULEr 48 87 10
SOLUtion TRIAl ENSEmble
mol1 EULEr 97 47 88
SOLUtion TRIAl ENSEmble
mol1 EULEr 25 15 79
When the rlist file is generated by Phaser, an additional keyword SCORE
appears on the end of the SOLUtion
TRIAl lines. This is the score from the rotation function. It is not
used by Phaser, but allows the user to keep track of the results.
SOLUtion SET
SOLUtion 6DIM ENSEmble
mol1 EULEr 17 20 32 FRACtional
0.12 0.05 0.74
SOLUtion TRIAl ENSEmble
mol1 EULEr 44 20 32 SCORe
34
SOLUtion TRIAl ENSEmble
mol1 EULEr 67 65 51 SCORe
30
SOLUtion TRIAl ENSEmble
mol1 EULEr 67 112 81 SCORe
28
SOLUtion TRIAl ENSEmble
mol1 EULEr 68 19 38 SCORe
27
If a degenerate translation function is performed, then a SOLUtion
TRIAl line is produced with the degenerate translation information present,
ready for performing the translation function on the third dimension.
SOLUtion TRIAl ENSEmble
mol1 EULEr 17 20 32 DEGEnerate
X FRACtional 0.05 0.74
2.4 How to Control Output
Selection of Peaks from Rotation and Translation Functions
The selection of peaks saved for output in the rotation and translation functions
can be done in four different ways. Peaks can either be selected by "PERCent",
"SIGma", "NUMber"
or "ALL", illustrated
below. "PERCent"
means that the cutoff value is the percentage of the top peak, where the value
of the top peak is defined as 100% and the value of the mean is defined as 0%.
"SIGma"
means that the cutoff value is the number of standard deviations (sigmas) over
the mean (otherwise known as the Z-score). "NUMber"
means that the cutoff value is the number of top peaks to select. "ALL"
mean that all peaks are selected.
The default is selection by "PERCent"
with the cutoff value set at 75%. This has the advantage that there are always
peaks output. If the solution is clear, and is a long way above the mean, then
only the clear solution(s) will be output, but if the distribution of peaks is
rather flat, then many peaks will be output for testing in the next part of the
molecular replacement procedure (e.g. many peaks selected from the rotation function
for testing with a translation function). If an absolute significance test is
required, then selection by "SIGma"
is more appropriate, although not all searches will produce output if the cutoff
value is too high (e.g. 5 sigma). If the distribution is very flat then it might
be better to select by "NUMber",
for example select the top 1000 peaks for testing in the translation function.
"ALL"
is for full 6 dimensional searches, where all the solutions from the rotation
function are output for testing in the translation function (although this should
never be necessary; it would be faster and probably just as likely to work if
the top 1000 peaks were used in this way).
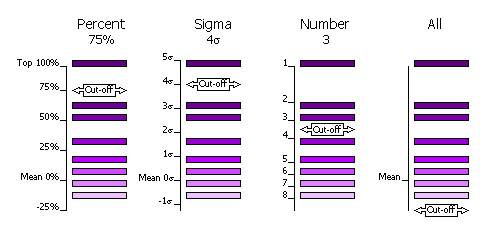
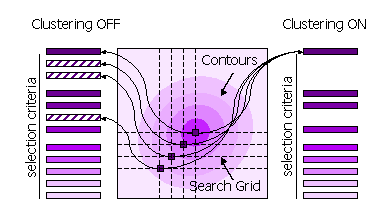
Peaks can also be clustered or not clustered prior to selection. If clustering
is off, then all high peaks on the search grid are selected. If clustering is
on, then points on the search grid with higher neighbouring points are removed
from the selection.
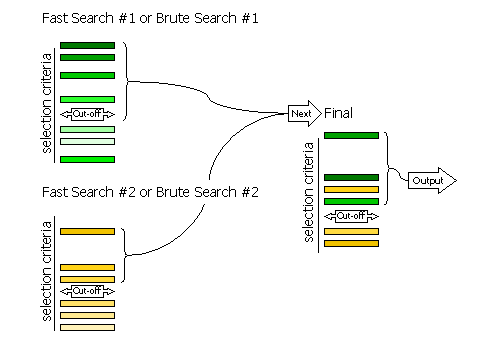
The selection of peaks is done in three stages for the fast rotation and fast
translation searches. The first stage is the selection of peaks from the fast
search that will be rescored with the full likelihood target. Rescoring with the
full likelihood target may change the order of the peaks and their significance.
The second stage is the selection of peaks from the rescoring to be saved and
combined with other searches performed in the same phaser job. The third stage
is the final selection of peaks from the merged list for output from the phaser
job. The selection of peaks to go into rescoring is controlled with the RESCORE
keyword, the selection of peaks saved from each separate search is controlled
with the SAVE keyword,
and the final selection is controlled with the FINAL
keyword.
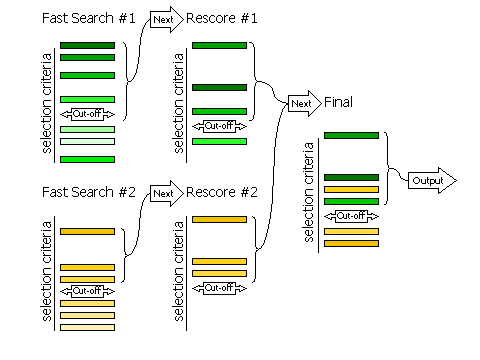
If RESCORE OFF
is requested (no rescoring of the fast search peaks is performed), or if
the brute rotation or translation searches are carried out, then the SAVE
keyword refers to the selection of peaks from the fast search (or brute search)
for merging in the final stage (the RESCORE
keyword is not used for selection in this case).
Map Coefficients
In the relevant modes (where HKLOut
ON is given as an optional keyword), Phaser produces an mtz file with
"SigmaA" type weighted Fourier map coefficients for producing electron density
maps for rebuilding.
| MTZ Column Labels |
Description |
| FWT |
PHWT |
Amplitude and phase for 2m|Fobs|-D|Fcalc| exp(i alpha-calc)
map |
| DELFWT |
PHDELWT |
Amplitude and phase for m|Fobs|-D|Fcalc| exp(i alpha-calc)
map |
| FOM |
|
m, analagous to the "Sim" weight, to estimate
the reliability of alpha-calc |
Additional Keywords
The output of Phaser can be controlled with the following keywords. The ROOT
keyword is not compulsory (the default root filename is PHASER), but should always
be given, so that your jobs have separate and meaningful output filenames.
OPTIONAL Keywords for Output
2.6 How to Run Phaser
Phaser runs in different modes, which perform Phaser's different functionalities,
such as rotation functions and translation functions. Some of the modes combine
the functionality of other modes to allow automatic structure solution (e.g. Molecular
Replacement Rotate Translate Pack), while others are basic modes with functionality
that may be useful outside of Phaser (e.g. Molecular Replacement
Anisotropy Correction).
The example scripts all refer to the tutorial test case, the crystal structure
of a hetero-dimer of beta-lactamase (BETA) and beta-lactamase inhibitor protein
(BLIP), both with molecular replacement models from crystal structures of the
individual BETA and BLIP components. The pdb and mtz files required for running
this test case are distributed with Phaser.
2.6.1 Automatic - Rotate Translate Pack
This mode combines the anisotropy correction, likelihood enhanced fast rotation
function, likelihood enhanced fast translation function, packing and refinement
modes for multiple search models to automatically solve a structure by molecular
replacement. Top solutions are output to the file FILEROOT.sol.
Example Script for Rotate Translate Pack
beta_blip_auto.com:
Find BETA and BLIP. The spacegroup recorded on the mtz file is P3221
but the other hand is also a possibility.
phaser ‹‹
eof
TITLe beta blip automatic
MODE MR_RTP
HKLIn beta_blip.mtz
LABIn F=Fobs SIGF=Sigma
ENSEmble beta PDBfile beta.pdb IDENtity 100
ENSEmble blip PDBfile blip.pdb IDENtity 100
COMPosition PROTein MW 28853 NUM 1 #beta
COMPosition PROTein MW 17522 NUM 1 #blip
SEARch ENSEmble beta NUM 1
SEARch ENSEmble blip NUM 1
PERMutations ON # not the default
SGALternative HAND # not the default
ROOT beta_blip_auto # not the default
eof
Relevant Keywords for Rotate Translate Pack
Keywords marked with an asterisk (*) are for "expert" use only
COMPULSORY Keywords for Molecular Replacement Rotate Translate Pack
MODE MR_RTP
HKLIn <FILENAME>
LABIn F = <F> SIGF
= <SIGF>
ENSEmble <MODLID> &
[{PDBfile
<PDBFILE> [RMS
| IDENtity] <NUM> {PDBfile
<PDBFILE> [RMS | IDENtity]
<NUM>}… BOXScale <BOXSCALE>
} |
{HKLIn <MTZFILE> F
= <F> P = <P> PROTein
MW <PMW> NUCLeic MW <NMW>
Extent <EX EY EZ> RMS
<RMS> CENTre <CX CY CZ> } ]
COMPosition &
[{[PROTein
| NUCLeic_acid] MW
<MW> NUMber
<NUM> } |
{ENSEmble
<MODLID> FRACtional <FRAC> } ]
SEARch ENSEmble
<MODLID> {OR
ENSEmble <MODLID>}… {NUMber
<NUM>}
OPTIONAL Keywords for Molecular Replacement Rotate Translate Pack
FINAl SELEct
[{SIGma <S>} | {NUMber
<N>} | {PERCent <P>} | ALL]
RESCore [ON|OFF]
RESCore SELEct [{SIGma
<S>} | {NUMber <N>} | {PERCent
<P>} | ALL]
RESCore CLUSter
[ON|OFF] {DUMP <NDUMP>} {LOG
[ON|OFF]}
SAMPling {ROTation <ROTSAMP>}
{TRAnslation <TRASAMP>}
SAVE SELEct [{SIGma
<S>} | {NUMber <N>} | {PERCent
<P>} | ALL]
SAVE CLUSter
[ON|OFF] {DUMP <NDUMP>} {LOG
[ON|OFF]}
SGALTernative [ALL | HAND
| {TEST <SG>}]
PACK <ALLOWED_CLASHES>
PERMutations [ON|OFF]
*CLMN {SPHEre <SPHERE>}
{LMINimum <LMIN>} {LMAXimum
<LMAX>}
*RESCore TARGet [WILSON |
RICE]
OPTIONAL Keywords for Data
RESOlution <HIRES>
<LORES>
SPACegroup <SG> {OR
<SG>}
*BINS {MINimum
<L>} {MAXimum <H>} {NUMber
<N>} {WIDTh <W>} {CUBIc
<A B C>}
*CELL <A B C ALPHA BETA GAMMA>
*OUTLier [{ON
<OUTLIER_PROB>} | OFF]
*Selection [ALL | CENTric
| ACENtric | {RANDom
<PC>}]
OPTIONAL Keywords for Models
SOLUtion
SET
<ANNOTATION>
SOLUtion
3DIM ENSEmble <MODLID> EULEr <A
B G> FIXR
SOLUtion
5DIM ENSEmble <MODLID> EULEr <A
B G> DEGEnerate [X|Y|Z] FRACtional
<U V> FIXR FIXT
SOLUtion
6DIM ENSEmble <MODLID> EULEr <A
B G> [ORTHogonal|FRACtional]
<X Y Z> FIXR FIXT
*BOXScale <BOXSCALE>
*COMPosition Water <PERCENT_WATER_SCAT>
*ENSEmble <MODLID> BINS
{MINimum <L>} {MAXimum
<H>} {NUMber <N>} {WIDTh
<W>} {CUBIc <A B C>}
*IPLN [1|2|3]
*SHANnon <SHARAT>
OPTIONAL Keywords for Output
2.6.2 Fast Rotation Function
This mode combines the anisotropy correction and likelihood enhanced fast rotation
function (2), optionally rescored with the full rotation
likelihood function (1), to find the orientation of a model in molecular replacement.
Top rotation solutions are output to the file FILEROOT.rlist for input to a translation
function. Top rotation solutions are also output to the file FILEROOT.sol.
Example Scripts for Fast Rotation Function
beta_frf.com:
Fast rotation function to find the orientation of BETA.
phaser ‹‹
eof
TITLe beta FRF
MODE MR_FRF
HKLIn beta_blip.mtz
LABIn F=Fobs SIGF=Sigma
ENSE beta PDBFile beta.pdb IDENtity 100
COMPosition PROTein MW 28853 NUM 1 #beta
COMPosition PROTein MW 17522 NUM 1 #blip
SEARCH ENSEmble beta
ROOT beta_frf
eof
blip_frf_with_beta.com:
Fast rotation function to find the orientation of BLIP knowing the position
and orientation of BETA, with the position and orientation of BETA input from
the command line.
phaser ‹‹
eof
TITLe blip FRF with beta RT
MODE MR_FRF
HKLIn beta_blip.mtz
LABIn F=Fobs SIGF=Sigma
ENSEmble beta PDBfile beta.pdb IDENtity 100
ENSEmble blip PDBfile blip.pdb IDENtity 100
COMPosition PROTein MW 28853 #beta
COMPosition PROTein MW 17522 #blip
SEARch ENSEmble blip
SOLUtion 6DIM ENSEmble beta EULEr 201 41 184 FRACtional -0.49408 -0.15571 -0.28148
ROOT blip_frf_with_beta
eof
blip_frf_with_beta_rot.com:
Fast rotation function to find the orientation of BLIP knowing only the
orientation of BETA, with the orientation of BETA input using the output solution
file from the beta_frf.com job
above.
phaser ‹‹
eof
TITLe blip FRF with beta R
MODE MR_FRF
HKLIn beta_blip.mtz
LABIn F=Fobs SIGF=Sigma
ENSEmble beta PDBfile beta.pdb IDENtity 100
ENSEmble blip PDBfile blip.pdb IDENtity 100
COMPosition PROTein MW 28853 NUM 1 #beta
COMPosition PROTein MW 17522 NUM 1 #blip
SEARch ENSEmble blip
@beta_frf.sol # solution file output by phaser
ROOT blip_frf_with_beta_rot
eof
Relevant Keywords for Fast Rotation Function
Keywords marked with an asterisk (*) are for "expert" use only
COMPULSORY Keywords for Molecular Replacement Fast Rotation Function
MODE MR_FRF
HKLIn <FILENAME>
LABIn F = <F> SIGF
= <SIGF>
ENSEmble <MODLID> &
[{PDBfile
<PDBFILE> [RMS
| IDENtity] <NUM> {PDBfile
<PDBFILE> [RMS | IDENtity]
<NUM>}… BOXScale <BOXSCALE>
} |
{HKLIn <MTZFILE> F
= <F> P = <P> PROTein
MW <PMW> NUCLeic_acid MW <NMW>
Extent <EX EY EZ> RMS
<RMS> CENTre <CX CY CZ> } ]
COMPosition &
[{[PROTein
| NUCLeic_acid] MW
<MW> NUMber
<NUM> } |
{ENSEmble
<MODLID> FRACtional <FRAC> } ]
SEARch ENSEmble
<MODLID> {OR
ENSEmble <MODLID>}… {NUMber
<NUM>}
OPTIONAL Keywords for Molecular Replacement Fast Rotation Function
FINAl SELEct
[{SIGma <S>} | {NUMber
<N>} | {PERCent <P>} | ALL]
RESCore [ON|OFF]
RESCore SELEct [{SIGma
<S>} | {NUMber <N>} | {PERCent
<P>} | ALL]
RESCore CLUSter
[ON|OFF] {DUMP <NDUMP>} {LOG
[ON|OFF]}
SAMPling {ROTAtion <ROTSAMP>}
SAVE SELEct [{SIGma
<S>} | {NUMber <N>} | {PERCent
<P>} | ALL]
SAVE CLUSter
[ON|OFF] {DUMP <NDUMP>} {LOG
[ON|OFF]}
*CLMN {SPHEre <SPHERE>}
{LMINimum <LMIN>} {LMAXimum
<LMAX>}
*RESCore TARGet [RICE | WILSON]
*TARGet [CROWTHER | LERF1 | LERF2]
OPTIONAL Keywords for Data
RESOlution <HIRES>
<LORES>
SPACegroup <SG> {OR
<SG>}
*BINS {MINimum
<L>} {MAXimum <H>} {NUMber
<N>} {WIDTh <W>} {CUBIc
<A B C>}
*CELL <A B C ALPHA BETA GAMMA>
*OUTLier [{ON
<OUTLIER_PROB>} | OFF]
*Selection [ALL | CENTric
| ACENtric | {RANDom
<PC>}]
OPTIONAL Keywords for Models
SOLUtion
SET
<ANNOTATION>
SOLUtion
3DIM ENSEmble <MODLID> EULEr <A
B G>
SOLUtion
5DIM ENSEmble <MODLID> EULEr <A
B G> DEGEnerate [X|Y|Z] FRACtional
<U V>
SOLUtion
6DIM ENSEmble <MODLID> EULEr <A
B G> [ORTHogonal|FRACtional]
<X Y Z>
OPTIONAL Keywords for Output
2.6.3 Brute Rotation Function
This mode combines the anisotropy correction and brute force likelihood rotation
function (1) to find the orientation of a model in molecular
replacement. Top rotation solutions are output to the file FILEROOT.rlist for
input to a translation function. Top rotation solutions are also output to the
file FILEROOT.sol.
Example Scripts for Brute Rotation Function
beta_brf.com:
Brute rotation function to find the orientation of BETA
phaser ‹‹
eof
TITLe beta BRF
MODE MR_BRF
HKLIn beta_blip.mtz
LABIn F=Fobs SIGF=Sigma
ENSEmble beta PDBfile beta.pdb IDENtity 100
COMPosition PROTein MW 28853 NUM 1 #beta
COMPosition PROTein MW 17522 NUM 1 #blip
SEARch ENSEmble beta
ROOT beta_brf
eof
beta_brf_around.com:
Brute rotation function to find the optimal orientation of BETA in a restricted
search range and on a fine grid around the position from the fast rotation search.
phaser ‹‹
eof
TITLe beta BRF fine sampling
MODE MR_BRF
HKLIn beta_blip.mtz
LABIn F=Fobs SIGF=Sigma
ENSEmble beta PDBfile beta.pdb IDENtity 100
ENSEmble blip PDBfile blip.pdb IDENtity 100
COMPosition PROTein MW 28853 NUM 1 #beta
COMPosition PROTein MW 17522 NUM 1 #blip
SEARch ENSEmble beta
ROTAte AROUnd EULEr 201 41 184 RANGE 10
SAMPling ROTAtion 0.5
XYZOut ON # not the default
TOPFiles 1 # not the default
ROOT beta_brf_around
eof
Relevant Keywords for Brute Rotation Function
Keywords marked with an asterisk (*) are for "expert" use only
COMPULSORY Keywords for Molecular Replacement Brute Rotation Function
MODE MR_BRF
HKLIn <FILENAME>
LABIn F = <F> SIGF
= <SIGF>
ENSEmble <MODLID> &
[{PDBfile
<PDBFILE> [RMS
| IDENtity] <NUM> {PDBfile
<PDBFILE> [RMS | IDENtity]
<NUM>}… BOXScale <BOXSCALE>
} |
{HKLIn <MTZFILE> F
= <F> P = <P> PROTein
MW <PMW> NUCLeic_acid MW <NMW>
Extent <EX EY EZ> RMS
<RMS> CENTre <CX CY CZ> } ]
COMPosition &
[{[PROTein
| NUCLeic_acid] MW
<MW> NUMber
<NUM> } |
{ENSEmble
<MODLID> FRACtional <FRAC> } ]
SEARch ENSEmble
<MODLID> {OR
ENSEmble <MODLID>}… {NUMber
<NUM>}
OPTIONAL Keywords for Molecular Replacement Brute Rotation Function
FINAl SELEct
[{SIGma <S>} | {NUMber
<N>} | {PERCent <P>} | ALL]
SAMPling {ROTAtion <ROTSAMP>}
SAVE SELEct [{SIGma
<S>} | {NUMber <N>} | {PERCent
<P>} | ALL]
SAVE CLUSter
[ON|OFF] {DUMP <NDUMP>} {LOG
[ON|OFF]}
Rotation FULL
Rotation AROUnd EULEr
<A B G> RANGe
<MAXE>
*TARGet [WILSON | RICE]
OPTIONAL Keywords for Data
RESOlution <HIRES>
<LORES>
SPACegroup <SG>
*BINS {MINimum
<L>} {MAXimum <H>} {NUMber
<N>} {WIDTh <W>} {CUBIc
<A B C>}
*CELL <A B C ALPHA BETA GAMMA>
*OUTLier [{ON
<OUTLIER_PROB>} | OFF]
*Selection [ALL | CENTric
| ACENtric | {RANDom
<PC>}]
OPTIONAL Keywords for Models
SOLUtion
SET
<ANNOTATION>
SOLUtion
3DIM ENSEmble <MODLID> EULEr <A
B G>
SOLUtion
5DIM ENSEmble <MODLID> EULEr <A
B G> DEGEnerate [X|Y|Z] FRACtional
<U V>
SOLUtion
6DIM ENSEmble <MODLID> EULEr <A
B G> [ORTHogonal|FRACtional]
<X Y Z>
*BOXScale
<BOXSCALE>
*COMPosition Water <PERCENT_WATER_SCAT>
*ENSEmble <MODLID> BINS
{MINimum <L>} {MAXimum
<H>} {NUMber <N>} {WIDTh
<W>} {CUBIc <A B C>}
*IPLN [1|2|3]
*SHANnon <SHARAT>
OPTIONAL Keywords for Output
2.6.4 Fast Translation Function
This mode combines the anisotropy correction and likelihood enhanced fast translation
function, optionally rescored by the full likelihood translation function, to
find the position of a previously oriented model in molecular replacement. Top
translation solutions are output to the file FILEROOT.sol.
Example Scripts for Fast Translation Function
beta_ftf.com:
The following script finds the position of BETA after the rotation function
has been run and the results output to the file beta_frf.rlist
phaser ‹‹
eof
TITLe beta FTF
MODE MR_FTF
HKLIn beta_blip.mtz
LABIn F=Fobs SIGF=Sigma
ENSEmble beta PDBFile beta.pdb IDENtity 100
ENSEmble blip PDBFile blip.pdb IDENtity 100
COMPosition PROTein MW 28853 NUM 1 #beta
COMPosition PROTein MW 17522 NUM 1 #blip
@beta_frf.rlist
ROOT beta_ftf
eof
blip_ftf_with_beta.com:
The following script finds the position of BLIP after the rotation function
has been run and the results output to the file blip_frf_with_beta.rlist,
which has the SOLUtion 6DIM keyword
input for BETA and the SOLUtion TRIAL
keyword input for the orientations to try for BLIP with the translation
function.
phaser ‹‹
eof
TITLe beta FTF
MODE MR_FTF
HKLIn beta_blip.mtz
LABIn F=Fobs SIGF=Sigma
ENSEmble beta PDBFile beta.pdb IDENtity 100
ENSEmble blip PDBFile blip.pdb IDENtity 100
COMPosition PROTein MW 28853 NUM 1 #beta
COMPosition PROTein MW 17522 NUM 1 #blip
@blip_frf_with_beta.rlist
ROOT blip_ftf_with_beta
eof
Relevant Keywords for Fast Translation Function
Keywords marked with an asterisk (*) are for "expert" use only
COMPULSORY Keywords for Molecular Replacement Fast Translation Function
MODE MR_FTP
HKLIn <FILENAME>
LABIn F = <F> SIGF
= <SIGF>
ENSEmble <MODLID> &
[{PDBfile
<PDBFILE> [RMS
| IDENtity] <NUM> {PDBfile
<PDBFILE> [RMS | IDENtity]
<NUM>}… BOXScale <BOXSCALE>
} |
{HKLIn <MTZFILE> F
= <F> P = <P> PROTein
MW <PMW> NUCLeic_acid MW <NMW>
Extent <EX EY EZ> RMS
<RMS> CENTre <CX CY CZ> } ]
COMPosition &
[{[PROTein
| NUCLeic_acid] MW
<MW> NUMber
<NUM> } |
{ENSEmble
<MODLID> FRACtional
<FRAC> } ]
SOLUtion
TRIAl ENSEmble <MODLID>
EULEr <A B C>
OPTIONAL Keywords for Molecular Replacement Fast Translation Function
FINAl SELEct
[{SIGma <S>} | {NUMber
<N>} | {PERCent <P>} | ALL]
RESCore [ON|OFF]
RESCore SELEct [{SIGma
<S>} | {NUMber <N>} | {PERCent
<P>} | ALL]
RESCore CLUSter
[ON|OFF] {DUMP <NDUMP>} {LOG
[ON|OFF]}
SAMPling {TRANslation <TRASAMP>}
SAVE SELEct [{SIGma
<S>} | {NUMber <N>} | {PERCent
<P>} | ALL]
SAVE CLUSter
[ON|OFF] {DUMP <NDUMP>} {LOG
[ON|OFF]}
SGALTernative [ALL | HAND
| {TEST <SG>}]
*RESCore TARGet [WILSON |
RICE]
*TARGet [CORRelation
| LETF1 ]
OPTIONAL Keywords for Data
RESOlution <HIRES>
<LORES>
SPACegroup <SG>
*BINS {MINimum
<L>} {MAXimum <H>} {NUMber
<N>} {WIDTh <W>} {CUBIc
<A B C>}
*CELL <A B C ALPHA BETA GAMMA>
*OUTLier [{ON
<OUTLIER_PROB>} | OFF]
*Selection [ALL | CENTric
| ACENtric | {RANDom
<PC>}]
OPTIONAL Keywords for Models
SOLUtion
SET
<ANNOTATION>
SOLUtion
3DIM ENSEmble <MODLID> EULEr <A
B G>
SOLUtion
5DIM ENSEmble <MODLID> EULEr <A
B G> DEGEnerate [X|Y|Z] FRACtional
<U V>
SOLUtion
6DIM ENSEmble <MODLID> EULEr <A
B G> [ORTHogonal|FRACtional]
<X Y Z>
*BOXScale
<BOXSCALE>
*COMPosition Water <PERCENT_WATER_SCAT>
*ENSEmble <MODLID> BINS
{MINimum <L>} {MAXimum
<H>} {NUMber <N>} {WIDTh
<W>} {CUBIc <A B C>}
*IPLN [1|2|3]
*SHANnon <SHARAT>
OPTIONAL Keywords for Output
2.6.5 Brute Translation Function
This mode combines the anisotropy correction and brute force likelihood translation
function (1) to find the position of a previously oriented
model in molecular replacement. Top translation solutions are output to the file
FILEROOT.sol.
Example Scripts for Brute Translation Function
beta_btf.com:
Brute Translation function to find the position of BETA after the rotation
function has been run
phaser ‹‹
eof
TITLe beta BTF
MODE MR_BTF
HKLIn beta_blip.mtz
LABIn F=Fobs SIGF=Sigma
ENSEmble beta PDBfile beta.pdb IDENtity 100
ENSEmble blip PDBfile blip.pdb IDENtity 100
COMPosition PROTein MW 28853 NUM 1 #beta
COMPosition PROTein MW 17522 NUM 1 #blip
@beta_frf.rlist
TRANslate AROUnd FRACtional POINt -0.49408 -0.15571 -0.28148 RANGe 5
ROOT beta_btf
eof
beta_btf_degen_x.com:
Brute Translation function to find the position of BETA degenerate in X
after the rotation function has been run
phaser ‹‹
eof
TITLe beta degenerate X
MODE MR_BTF
HKLIn beta_blip.mtz
LABIn F=Fobs SIGF=Sigma
ENSEmble beta PDBfile beta.pdb IDENtity 100
ENSEmble blip PDBfile blip.pdb IDENtity 100
COMPosition PROTein MW 28853 NUM 1 #beta
COMPosition PROTein MW 17522 NUM 1 #blip
@beta_frf.rlist
TRANslate DEGEnerate X
ROOT beta_btf_degen_x
eof
Relevant Keywords for Brute Translation Function
Keywords marked with an asterisk (*) are for "expert" use only
COMPULSORY Keywords for Molecular Replacement Brute Translation Function
MODE MR_BTF
HKLIn <FILENAME>
LABIn F = <F> SIGF
= <SIGF>
ENSEmble <MODLID> &
[{PDBfile
<PDBFILE> [RMS
| IDENtity] <NUM> {PDBfile
<PDBFILE> [RMS | IDENtity]
<NUM>}… BOXScale <BOXSCALE>
} |
{HKLIn <MTZFILE> F
= <F> P = <P> PROTein
MW <PMW> NUCLeic_acid MW <NMW>
Extent <EX EY EZ> RMS
<RMS> CENTre <CX CY CZ> } ]
COMPosition &
[{[PROTein
| NUCLeic_acid] MW
<MW> NUMber
<NUM> } |
{ENSEmble
<MODLID> FRACtional
<FRAC> } ]
SOLUtion
TRIAl ENSEmble <MODLID> EULEr <A
B C>
OPTIONAL Keywords for Molecular Replacement Brute Translation Function
FINAl SELEct
[{SIGma <S>} | {NUMber
<N>} | {PERCent <P>} | ALL]
SAMPling {TRANslation <TRASAMP>}
SAVE SELEct [{SIGma
<S>} | {NUMber <N>} | {PERCent
<P>} | ALL]
SAVE CLUSter
[ON|OFF] {DUMP <NDUMP>} {LOG
[ON|OFF]}
SGALTernative [ALL | HAND
| {TEST <SG>}]
TRANslate
FULL
TRANslate LINE [ORTHogonal
| FRACtional] STARt
<XS YS ZS> END <XE YE ZE>
TRANslate REGIon [ORTHogonal
| FRACtional] STARt
<XS YS ZS> END <XE YE ZE>
TRANslate AROUnd [ORTHogonal
| FRACtional] POINt
<X Y Z> RANGe <RANGE>
TRANslate DEGEnerate [X|Y|Z]
TARGet [WILSON | RICE]
OPTIONAL Keywords for Data
RESOlution <HIRES>
<LORES>
SPACegroup <SG>
*BINS {MINimum
<L>} {MAXimum <H>} {NUMber
<N>} {WIDTh <W>} {CUBIc
<A B C>}
*CELL <A B C ALPHA BETA GAMMA>
*OUTLier [{ON
<OUTLIER_PROB>} | OFF]
*Selection [ALL | CENTric
| ACENtric | {RANDom
<PC>}]
OPTIONAL Keywords for Models
SOLUtion
SET
<ANNOTATION>
SOLUtion
3DIM ENSEmble <MODLID> EULEr <A
B G>
SOLUtion
5DIM ENSEmble <MODLID> EULEr <A
B G> DEGEnerate [X|Y|Z] FRACtional
<U V>
SOLUtion
6DIM ENSEmble <MODLID> EULEr <A
B G> [ORTHogonal|FRACtional]
<X Y Z>
*BOXScale
<BOXSCALE>
*COMPosition Water <PERCENT_WATER_SCAT>
*ENSEmble <MODLID> BINS
{MINimum <L>} {MAXimum
<H>} {NUMber <N>} {WIDTh
<W>} {CUBIc <A B C>}
*IPLN [1|2|3]
*SHANnon <SHARAT>
OPTIONAL Keywords for Output
2.6.6 Refinement and Phasing
This mode combines the anisotropy correction and refinement against the likelihood
function (1) to optimize full or partial molecular replacement
solutions and phase the data. At the end of refinement, the list of solutions
is checked for duplicates, which are pruned. Refined solutions are output to the
file FILEROOT.sol.
Example Script for Refinement and Phasing
beta_blip_rnp.com:
Refines the set of solutions in the file beta_blip.sol
phaser ‹‹
eof
TITLe beta blip rigid body refinement
MODE MR_RNP
HKLIn beta_blip.mtz
LABIn F=Fobs SIGF=Sigma
ENSEmble beta PDBfile beta.pdb IDENtity 100
ENSEmble blip PDBfile blip.pdb IDENtity 100
COMPosition PROTein MW 28853 NUM 1 #beta
COMPosition PROTein MW 17522 NUM 1 #blip
ROOT beta_blip_rnp # not the default
HKLOut OFF # not the default
XYZOut OFF # not the default
@beta_blip.sol
eof
Relevant Keywords for Refinement and Phasing
Keywords marked with an asterisk (*) are for "expert" use only
COMPULSORY Keywords for Molecular Replacement Refinement and Phasing
MODE MR_RNP
HKLIn <FILENAME>
LABIn F = <F> SIGF
= <SIGF>
ENSEmble <MODLID> &
[{PDBfile
<PDBFILE> [RMS
| IDENtity] <NUM> {PDBfile
<PDBFILE> [RMS | IDENtity]
<NUM>}… BOXScale <BOXSCALE>
} |
{HKLIn <MTZFILE> F
= <F> P = <P> PROTein
MW <PMW> NUCLeic_acid MW <NMW>
Extent <EX EY EZ> RMS
<RMS> CENTre <CX CY CZ> } ]
COMPosition &
[{[PROTein
| NUCLeic_acid] MW
<MW> NUMber
<NUM> } |
{ENSEmble
<MODLID> FRACtional <FRAC> } ]
SOLUtion &
[{SET
<ANNOTATION> |
{3DIM ENSEmble <MODLID> EULEr
<A B G> FIXR} |
{5DIM ENSEmble <MODLID> EULEr
<A B G> DEGEnerate [X|Y|Z] FRACtional
<U V> FIXR FIXT } |
{6DIM
ENSEmble <MODLID> EULEr
<A B G> [ORTHogonal|FRACtional]
<X Y Z> FIXR
FIXT} ]
OPTIONAL Keywords for Molecular Replacement Refinement and Phasing
*MRMAcrocycle
ROTAtion [ON|OFF] TRANslation
[ON|OFF] NCYCle <NCYC>
TARGet [WILSON | RICE] MINImizer
<MINIMIZER>
*TARGet [WILSON
| RICE]
OPTIONAL Keywords for Data
RESOlution <HIRES>
<LORES>
SPACegroup <SG>
*BINS {MINimum
<L>} {MAXimum <H>} {NUMber
<N>} {WIDTh <W>} {CUBIc
<A B C>}
*CELL <A B C ALPHA BETA GAMMA>
*OUTLier [{ON <OUTLIER_PROB>} | OFF]
*Selection [ALL | CENTric
| ACENtric | {RANDom
<PC>}]
OPTIONAL Keywords for Models
OPTIONAL Keywords for Output
2.6.7 Log-Likelihood Gain
This mode combines the anisotropy correction and the likelihood function (1)
to calculate the log-likelihood gain for full or partial molecular replacement
solutions. Solutions are output to the file FILEROOT.sol.
Example Script for Log-Likelihood Gain
beta_blip_llg.com:
Rescore the solutions using a different resolution range of data and a
different spacegroup
phaser ‹‹
eof
TITLe beta blip solution 6A P3121
MODE MR_LLG
HKLIn beta_blip.mtz
LABIn F=F SIGF = SIGF
ENSEmble beta PDBfile beta.pdb IDENtity 100
ENSEmble blip PDBfile blip.pdb IDENtity 100
COMPosition PROTein MW 28853 NUM 1 #beta
COMPosition PROTein MW 17522 NUM 1 #blip
ROOT beta_blip_llg # not the default
RESOlution 6.0
SPACegroup P 31 2 1
@beta_blip.sol
eof
Relevant Keywords for Log-Likelihood Gain
Keywords marked with an asterisk (*) are for "expert" use only
COMPULSORY Keywords for Molecular Replacement Log-Likelihood Gain
MODE MR_LLG
HKLIn <FILENAME>
LABIn F = <F> SIGF
= <SIGF>
ENSEmble <MODLID> &
[{PDBfile
<PDBFILE> [RMS
| IDENtity] <NUM> {PDBfile
<PDBFILE> [RMS | IDENtity]
<NUM>}… BOXScale <BOXSCALE>
} |
{HKLIn <MTZFILE> F
= <F> P = <P> PROTein
MW <PMW> NUCLeic_acid MW <NMW>
Extent <EX EY EZ> RMS
<RMS> CENTre <CX CY CZ> } ]
COMPosition &
[{[PROTein
| NUCLeic_acid] MW
<MW> NUMber
<NUM> } |
{ENSEmble
<MODLID> FRACtional <FRAC> } ]
SOLUtion &
[{SET
<ANNOTATION> |
{3DIM ENSEmble <MODLID> EULEr
<A B G> } |
{5DIM ENSEmble <MODLID> EULEr
<A B G> DEGEnerate [X|Y|Z] FRACtional
<U V> } |
{6DIM
ENSEmble <MODLID> EULEr
<A B G> [ORTHogonal|FRACtional]
<X Y Z>} ]
OPTIONAL Keywords for Molecular Replacement Log-Likelihood Gain
OPTIONAL Keywords for Data
RESOlution <HIRES>
<LORES>
SPACegroup <SG>
*BINS {MINimum
<L>} {MAXimum <H>} {NUMber
<N>} {WIDTh <W>} {CUBIc
<A B C>}
*CELL <A B C ALPHA BETA GAMMA>
*OUTLier [{ON
<OUTLIER_PROB>} | OFF]
*Selection [ALL | CENTric
| ACENtric | {RANDom
<PC>}]
OPTIONAL Keywords for Models
OPTIONAL Keywords for Output
2.6.8 Packing
This mode determines whether molecular replacement solutions pack in the unit
cell. Solutions that pack are output to the file FILEROOT.sol.
Example Script for Packing
beta_blip_pak.com:
Determine whether a set of molecular replacement solutions pack in the
unit cell
phaser ‹‹
eof
TITLe beta blip packing check
MODE MR_PAK
HKLIn beta_blip.mtz
LABIn F=F SIGF=SIGF
ENSEmble beta PDBfile beta.pdb IDENtity 100
ENSEmble blip PDBfile blip.pdb IDENtity 100
COMPosition PROTein MW 28853 NUM 1 #beta
COMPosition PROTein MW 17522 NUM 1 #blip
ROOT beta_blip_pak # not the default
PACK 1 # not the default
@beta_blip.sol
eof
Relevant Keywords for Packing
Keywords marked with an asterisk (*) are for "expert" use only
COMPULSORY Keywords for Molecular Replacement Packing
MODE MR_PAK
HKLIn <FILENAME>
LABIn F = <F> SIGF
= <SIGF>
ENSEmble <MODLID> &
PDBfile
<PDBFILE> [RMS
| IDENtity] <NUM> {PDBfile
<PDBFILE> [RMS | IDENtity]
<NUM>}… BOXScale <BOXSCALE>
SOLUtion &
[{SET
<ANNOTATION> |
{3DIM ENSEmble <MODLID> EULEr
<A B G> } |
{5DIM ENSEmble <MODLID> EULEr
<A B G> DEGEnerate [X|Y|Z] FRACtional
<U V> } |
{6DIM
ENSEmble <MODLID> EULEr
<A B G> [ORTHogonal|FRACtional]
<X Y Z>} ]
OPTIONAL Keywords for Molecular Replacement Packing
OPTIONAL Keywords for Data
OPTIONAL Keywords for Output
2.6.9 Anisotropy Correction
This mode corrects the experimental data for anisotropy. Data (amplitude and associated
sigma) are corrected for anisotropy and output to FILEROOT.mtz with column label
set to the input column label with the addition of _ISO.
Example Script for Anisotropy Correction
beta_blip_ano.com:
Phase a molecular replacement solution only
phaser ‹‹
eof
TITLe beta blip data correction
MODE MR_ANO
HKLIn beta_blip.mtz
LABIn F=Fobs SIGF=Sigma
ROOT beta_blip_ano # not the default
eof
Relevant Keywords for Anisotropy Correction
Keywords marked with an asterisk (*) are for "expert" use only
COMPULSORY Keywords for Anisotropy Correction
OPTIONAL Keywords for Anisotropy Correction
*SNMAcrocycle ANISotropic
[ON|OFF] BINS [ON|OFF] SOLK
[ON|OFF] SOLB [ON|OFF] NCYCle
<NCYC> MINImizer <MINIMIZER>
OPTIONAL Keywords for Data
OPTIONAL Keywords for Output
2.7. How to know whether Phaser has solved it
By default, phaser selects solutions over 75% of the the difference between the
top solution and the mean. Ideally, only the number of solutions you are expecting
should be selected by this criteria, but if the signal-to-noise of your search
is low, there will be noise peaks in this selection also. For a translation function
the correct solution will generally have a Z-score (number of standard deviations
above the mean value) over 5 and be well separated from the rest of the solutions.
For a rotation function, the correct solution may be in the list with a Z-score
under 4, and will not be found until a translation function is performed and picks
out the correct solution.
|
Z-score
|
Have I solved it?
|
|
less than 5
|
no
|
|
5 - 6
|
unlikely
|
|
6 - 7
|
possibly
|
|
7 - 8
|
probably
|
|
more than 8
|
definitely
|
Of course, there will always be exceptions!
2.8. What to do in difficult cases
Not every structure can be solved by molecular replacement, but the right strategy
can push the limits. What to do when the default jobs fail depends on why your
structure is difficult.
Flexible structure
The relative orientations of the domains may be different in your crystal than
in the model. If that may be the case, break the model into separate PDB files
containing rigid-body units, enter these as separate ensembles, and search for
them separately. If you find a convincing solution for one domain, but fail to
find a solution for the next domain, you can take advantage of the knowledge that
its orientation is likely to be similar to that of the first domain. The ROTAte
AROUnd option of the brute rotation search can be used to restrict the
search to orientations within, say, 30 degrees of that of the known domain. Allow
for close approach of the domains by increasing the allowed clashes with the PACK
keyword by, say, 1 for each domain break that you introduce.
Poor or incomplete model
Signal-to-noise is reduced by coordinate errors or incompleteness of the model.
Since the rotation search has lower signal to begin with than the translation
search, it is usually more severely affected. For this reason, it can be very
useful to use a subsequent translation search as a way to choose among many (say
1000) orientations. Try increasing the number of clustered orientations in one
job using the keyword FINAl,
e.g. FINAl SELEct PERCent
65. If that fails, try turning off the clustering feature in the save step
(SAVE CLUSter OFF),
because the correct orientation may sit on the shoulder of a peak in the rotation
function.
High degree of non-crystallographic symmetry
If there are clear peaks in the self-rotation function, you can expect orientations
to be related by this known NCS. Methods to automatically use such information
will be implemented in a future version of Phaser. In the meantime, you can
work out for yourself the orientations that would be consistent with NCS and
use the ROTAte
AROUnd option to sample similar orientations.
Alternatively, you may have an oligomeric model and expect similar NCS in the
crystal. First search with the oligomeric model; if this fails, search with
a monomer. If that succeeds, you can again use the ROTAte
AROUnd option to force a subsequent monomer to adopt an orientation
similar to the one you expect.
Pseudo-translational non-crystallographic symmetry
It is frequently the case that crystallographic and non-crystallographic rotational
symmetry axes are parallel. The combination generates translational NCS, in which
more than one unique copy of the molecule is found in the same orientation in
the crystal. This can be recognized by the presence of large non-origin peaks
in the native Patterson map. If one copy of the search model can be found, then
the translational NCS tells you where to place another copy. Unfortunately, the
presence of translational NCS can make it difficult to solve a structure using
Phaser, because the current likelihood targets do not account for the statistical
effects of NCS.
3. Preprocessor
Preprocessor commands (@
# & END STOP QUIT EXIT KILL) may be used in the keyword input to incorporate
files, add comments or allow line continuation.
- @<filename>
includes a file in the input stream
- All characters on a line after a hash (#)
character are ignored
- Line continuation with the ampersand (&)
character
- END STOP QUIT EXIT KILL
or "eof" from a command file ends the input and starts
Phaser
4. Keywords
Phaser can be controlled using keyword input. Not all keywords are relevant for
all modes of operation (the list of relevant keywords for each mode is given with
each mode above). Some keywords are only for single use, others have meaning when
used more than once. The input values of many parameters are constrained to physically
meaningful values. All non-compulsory parameters have defaults. Keywords marked
with an asterisk are for "expert" use only, or use for development.
- *BINS
{MINimum
<L>} {MAXimum <H>}
{NUMber <N>} {WIDTh
<W>} {CUBIc <A B C>}
- The binning of the data. L = minimum number of bins, H = maximum number
of bins, N = number of bins, W = width of the bins in number of reflections,
A B C are the coefficients for the binning function A(S*S*S)+B(S*S)+CS where
S = (1/resolution). If N is given then the values of L and H are ignored.
Single Use
Constraints L,H,N,W=integer(+)
CUBIc restricted to monotonically increasing: Either (a) AC
>0,BC
>0,C
>0 or (b)
A=B=0 or (c) A=0 or
(d) B=0
Default BINS
MINimum 6 MAXimum 50 WIDTh
1000 CUBIc 0 1 0
- *BOXScale
<BOXSCALE>
- Scale for box for calculating structure factors. The ensembles are put
in a box equal to (extent of molecule)*BOXSCALE.
This BOXSCALE applies to all
ensembles, except those for which BOXSCALE
has been set individually using the ENSEmble
keyword.
Single Use
Constraints BOXSCALE
>2.4
Default BOXScale
4
- *CELL
<A B C ALPHA BETA GAMMA>
- Unit cell dimensions
Single Use
Constraints A>0,B>0,C>0,ALPHA>0,BETA>0,GAMMA>0
Default Cell read from MTZ file
- *CLMN
{SPHEre <SPHERE>} {LMINimum
<LMIN>} {LMAXimum <LMAX>}
- The radii or L values for the decomposition of the Patterson in Ångstroms.
Single Use
Constraints SPHERE>5,LMIN>0,LMAX>LMIN
Default CLMN
SPHEre <2*geometric mean radius of Ensemble> LMIN
2
- COMPosition
[PROTein | NUCLeic_acid]
MW <MW> NUMber
<NUM>
- Composition of the crystals. The number of copies NUMber of molecular weight
MW of protein or nucleic acid in the asymmetric unit.
Multiple Use
Constraints MW>0,NUMber=integer(+)
Default None for MW,
compulsory when required. NUMber
defaults to 1.
- COMPosition
ENSEmble <MODLID> FRACtional
<FRAC>
- Alternative way of defining composition. Fraction scattering is entered
explicitly for each ENSEmble .
Multiple Use
Constraints 0<FRAC<=1
Default None, compulsory when required
- *COMPosition
WATEr <WATER>
- Fraction of molecular weight added for ordered water content
Single Use
Constraints WATER=%
Default COMPosition
Water 5
- ENSEmble
<MODLID> PDBfile <PDBFILE> [RMS
| IDENtity] <NUM> {PDBfile
<PDBFILE> [RMS | IDENtity]
<NUM>}… BOXScale <BOXSCALE>
- The names of the PDB files used to build the ENSEmble
, and either the expected RMS deviation of the coordinates to the "real"
structure or the percent sequence identity with the real sequence.
Multiple Use
Constraints BOXSCALE
>0
Default None, compulsory when required
- ENSEmble
<MODLID> HKLIn <MTZFILE> F
= <F> P = <P> PROTein
MW <PMW> NUCLeic_acid MW <NMW>
Extent <EX EY EZ> RMS
<RMS> CENTre <CX CY CZ>
- An ENSEmble defined from a map (via an mtz file). The molecular weight
of the object the map represents is required for scaling, as is the RMS. Molecular
replacement translation functions will be given with respect to the centre
CX CY CZ
Multiple Use
Default None, compulsory when required
- *ENSEmble
<MODLID> BINS {MINimum
<L>} {MAXimum <H>} {NUMber
<N>} {WIDTh <W>} {CUBIc
<A B C>}
- Bins for the MODLID
Multiple Use
Constraints L,H,N,W
= integer(+) CUBIc restricted to monotonically increasing: Either (a)
AC >0,BC
>0,C >0
or (b) A=B=0 or (c)
A=0 or (d) B=0
Default ENSEmble
<MODLID> BINS MINimum 6 MAXimum
200 WIDTh 1000 CUBIc
0 1 0
- *ENSEmble
<MODLID> WRITE [INTErpolation | LERF |
CROWther] HKLOut <HKLOUT> E=<E>
P=<P> V=<V>
- Multiple Use
Constraints None
Default None, compulsory when required
- FINAl
SELEct [{SIGma <S>}
| {NUMber <N>} | {PERCent
<P>} | ALL]
- Final selection criteria for peaks. If no criteria are given for saving
or rescoring steps, then the criteria given for final selection is used as
the criteria for the saving and rescoring steps also.
Single Use
Constraints S>0,N=integer(+),P=%
Default FINAl
SELEct PERCent 75
- HKLIn
<FILENAME>
- The mtz file containing the data
Single Use
Default None, compulsory when required
- HKLOut
[ON|OFF]
- Flags for output of an mtz file containing the phasing information
Single Use
Default
HKLOut ON
- *IPLN
[1|2|3]
- Molecular transform interpolation level: 1= one point interpolation, 2 =
four point interpolation, 3 = eight point interpolation.
Single Use
Default IPLN
2
- LABIn
F = <F> SIGF =
<SIGF>
- Columns in mtz file. F must be given. SIGF should be given but is optional.
Single Use
Default None, compulsory when required
- MODE
[MR_RTP | MR_FRF | MR_FTF | MR_BRF | MR_BTF | MR_RNP | MR_LLG | MR_PAK | MR_ANO]
- The mode of operation of Phaser
Single Use
Default None, compulsory
- *MRMAcrocycle
ROTAtion [ON|OFF] TRANnslation
[ON|OFF] NCYCle <NCYC> TARGet
[WILSON | RICE] MINImizer <MINIMIZER>
- Molecular replacement refinement macrocycle. The macrocycles are performed
in the order that they are entered
Multiple Use
Default MRMAcrocycle
ROTAtion ON TRANslation
ON NCYCle 10 TARGet
RICE MINImizer BFGS
-
- *NORMalisation
{BINS <B1 B2 B3 …>} {ANISotropic
<HH KK LL HK HL KL>} {ISOB <ISOB>}
{SOLK <SOLK>} {SOLB
<SOLB>} FIXB FIXA FIXS FIXK
- Normalisation parameters for the data. Normalisation factor for reflection
hkl in bin i is given by SigmaN = Bi*(1-SOLK*exp(-SOLB))* exp(-(HH*h*h + KK*k*k
+ LL*l*l + HK*h*k + HL*h*l + KL*k*l))
Single Use
Default Calculated by Phaser
- *OUTLier
[{ON <OUTLIER_PROB>} | OFF]
- Control of the large unlikely E-value rejection. Outliers with a probability
less than OUTLIER_PROB are rejected.
Single Use
Default OUTLier
ON 0.000001
- PACK
<ALLOWED_CLASHES>
- NUMber of C-alpha atoms that can clash within 2A.
Single Use
Default PACK
0
- PERMutations
[ON|OFF]
- Toggle for whether the order of the search set is to be permuted.
Single Use
Default PERMutations
OFF
- *REFLection
<H K L FMEAN SIGFMEAN>
- Reflection data input manually.
Multiple Use
Default None
- RESCore
[ON|OFF]
- Toggle for rescoring of fast search peaks
Single Use
Default RESCore
ON
- RESCore
SELEct [{SIGma <S>}
| {NUMber <N>} | {PERCent
<P>} | ALL]
- Selection criteria for peaks to be rescored.
Single Use
Constraints S>0,N=integer(+),P=%
Default RESCore
SELEct PERCent 67.5
- RESCore CLUSter
[ON|OFF] {DUMP <NDUMP>} {LOG
[ON|OFF]}
- CLUS ON or OFF selects whether raw or clustered peaks are to be used in
the rescoring. If clustered peaks are used, then NDUMP raw peaks will be dumped
to the output. If clustered peaks are not used, then you may still perform
the clustering and log the results to the log file (this may be time consuming
if a large number of peaks are selected for clustering).
Single Use
Constraints NDUMP>0
- Default RESCore
CLUSter OFF LOG
ON
- *RESCore
TARGet [WILSON | RICE]
- Target function for the rescoring
Single Use
Default RESCore
TARGet RICE
- RESOlution
<HIRES> <LORES>
- Resolution range in Angstroms. If only one limit is given, it is the high
resolution limit; otherwise the limits can be in either order.
Single Use
Constraints HIRES>0,LORES
>0
Default Resolution range set to accommodate all input
reflections
- ROOT
<FILEROOT>
- Root filename for output files (e.g. FILEROOT.log)
Single Use
Default ROOT
PHASER
- ROTAte
FULL
- Sample all unique angles
- ROTAte
AROUnd EULEr <A B G> RANGe
<RANGE>
- Restrict the search to the region of
+/- RANGE degrees around orientation <A
B G>
Single Use
Constraints RANGE>0
Default ROTAte
FULL
- SAMPling
{ROTAtion <ROTSAMP>} {TRANslation
<TRASAMP>}
- Sampling of search given in degrees for a rotation search and Angstroms
for a translation search.
Single Use
Constraints ROTSAMP>0,
TRASAMP>0
Default SAMPling
ROTSAMP
<2*atan(dmin/(4*geometric mean radius))> TRASAMP
<dmin/5> for brute force search, <dmin/3>
for fast translation search
- SAVE
SELEct [{SIGma <S>}
| {NUMber <N>} | {PERCent
<P>} | ALL]
- Peaks satisfying selection criteria are saved. If no criteria are given
for the rescoring step, then the criteria given for saving is used as the
criteria for the rescoring step also.
Single Use
Constraints S>0,N=integer(+),P=%
Default SAVE
SELEct PERCent 75
- SAVE CLUSter
[ON|OFF] {DUMP <NDUMP>} {LOG
[ON|OFF]}
- CLUSter ON selects clustered
peaks for saving. If clustered peaks are used, then NDUMP raw peaks will be
dumped to the output. CLUSter OFF
selects raw peaks for saving. If clustered peaks are not used, then
you may still perform the clustering and log the results to the log file (this
may be time consuming).
Single Use
Constraints NDUMP>0
Default SAVE
CLUSter ON DUMP
20
- *SCRIpt
[ON|OFF]
- Write Phaser script file
Single Use
Default SCRIpt
ON
- SEARch
ENSEmble <MODLID> {OR
ENSEmble <MODLID>}… {NUMber
<NUM>}
- The ENSEmble to be searched for in a rotation
search or an automatic search. When multiple ensembles are given using the OR keyword, the search
is performed for each ENSEmble in turn. The final
results are the best of all the searches (controlled with the FINAL keyword).
When the keyword is entered multiple times in the MR_RTP mode, each SEARCH
keyword refers to a new component of the structure. If the component is present
multiple times the sub-keyword NUMber can be used (rather than entering the
same SEARCH keyword NUMber times). If the MR_RTP mode is being used with a
fixed partial solution, only enter SEARCH keywords (or associated NUMbers)
for the components that remain to be found.
Multiple Use
Constraints ENSEmble MODLID must be defined
Default None, compulsory when required
- *SELEct
[ALL | CENTric |
ACENtric | {RANDom
<PC>}]
- Select subset of reflections, either all reflections, centric only, acentric
only, or a random percent of reflections.
Single Use
Constraints PC=%
Default SELEct
ALL
- SGALternative
[ALL
| HAND | {TEST <SG>}]
- Alternative space groups to test in the translation function. All tests
all possible space groups, hand tests the given spacegroup and its enantiomorph
and <SG> tests the give space group.
Multiple Use
Default None
- *SHANnon
<SHARAT>
- Shannon sampling given by (2*SHARAT)
for the Ensemble maps. Increase SHARAT
to 2 to sharpen the sampling.
Single Use
Constraints SHARAT>1.1
Default SHANnon
1.5
- *SNMAcrocycle
ANISotropic [ON|OFF] BINS
[ON|OFF] SOLK [ON|OFF] SOLB
[ON|OFF] NCYCle <NCYC> MINImizer
<MINIMIZER>
- Macrocycles for the refinement of SigmaN in the anisotropy correction
Multiple Use
Constraints NCYC=integer(+)
Default SNMAcrocycle
ANISotropic ON BINS
ON SOLK OFF SOLB
OFF NCYCle 50 MINImizer
BFGS
- SOLUtion
SET <ANNOTATION>
- STARt new set of solutions
Multiple Use
- SOLUtion 3DIM
ENSEmble <MODLID> EULEr <A B
G> FIXR
- Rotation only solution. Use this keyword if only the orientation in 3 dimensions
is known. This keyword is repeated for each case. A B G are the Euler angles
in degrees.
Multiple Use
- SOLUtion
5DIM ENSEmble <MODLID> EULEr <A
B G> DEGEnerate [X|Y|Z] FRACtional
<U V> FIXR FIXT
- Use this keyword if the orientation in 3 dimensions and and the position
of the MODLID in only 2 dimensions is known. This keyword is repeated for
each case. A B G are the Euler angles in degrees. The keywords [X|Y|Z]specify
the degenerate direction and U V are the translation in the other two directions.
Multiple Use
- SOLUtion
6DIM ENSEmble <MODLID> EULEr <A
B G> [ORTHogonal|FRACtional]
<X Y Z> FIXR FIXT
- This keyword is repeated for each known position and orientation of a ENSEmble
ID. A B G are the Euler angles and X Y Z are the translation.
Multiple Use
- SOLUtion
TRIAl ENSEmble <MODLID> EULEr <A
B G> {DEGEnerate [X|Y|Z] FRACtional
<U V> } {SCORe <score>}
- Rotation List for translation function
Multiple Use
Default none, compulsory when required
- SOLUtion
SPACegroup<SG>
- Spacegroup for this solution
Multiple Use
Default Spacegroup for data
- SPACegroup
<SG>
- Spacegroup may be altered from the one on the MTZ file to a spacegroup in
the same point group. The spacegroup name or number can be given e.g. P 21
21 21 or 19
Single Use
Default read from MTZ file
- *SUITe
[CCP4 | PHENIX | CIMR]
- Switch output to match the style of ccp4, phenix and cimr (development).
Single Use
Default SUITe
CCP4
- *TARGet
[WILSON | RICE | LERF1 | LERF2 | CROWTHER | CORRelation | LETF1 ]
- Target function for mode
Single Use
Default for fast rotation function TARGet
LERF1
Default for fast translation function TARGet
LETF1
Default for all other modes TARGet
RICE
- TITLe
<TITLE>
- Title for job
Single Use
Default TITLe
[no title given]
- TOPFiles
<NUM>
- Number of top pdbfiles or mtzfiles to write to output.
Single Use
Constraints
NPDB=integer(+)
Default TOPFiles
1
- TRANslate
FULL
- Search volume for brute force translation function. Cheshire cell or Primitive
cell volume.
- TRANslate
LINE [ORTHogonal|FRACtional]
STARt <XS YS ZS> END
<XE YE ZE>
- Search volume for brute force translation function. Search along line.
- TRANslate
REGIon [ORTHogonal|FRACtional]
STARt <XS YS ZS> END
<XE YE ZE>
- Search volume for brute force translation function. Search region.
- TRANslate
AROUnd [ORTHogonal|FRACtional]
POINt <X Y Z> RANGe
<RANGE>
- Search volume for brute force translation function. Search within +/-
RANGE Angstroms (not fractional coordinates, even if the search point
is given as fractional coordinates) of a point <X
Y Z>.
- TRANslate
DEGEnerate [X|Y|Z]
- Search volume for brute force translation function. The search volume is
the plane perpendicular to the direction of the search.
Single Use
Default TRANslate
FULL
- VERBose
[{ON EXTRA} |OFF]
- Toggle to send verbose output to log file. If ON or OFF are not specified,
verbose is switched ON. If EXTRA is ON, then extra verbose information is
logged.
Single Use
Default VERBose
OFF
- XYZOut
[ON|OFF]
- Toggle for output coordinate files.
Single Use
Constraints
NPDB=integer(+)
Default Rotation functions XYZOut
OFF All
other (relevant) modes
XYZOut ON
Constraints
X=integer(+)
If X is a positive integer,
stored value = X
Else Input Error
X=%
If 1<X<=100, stored
value = X
Else If X<1, stored value
= X*100
Else Input Error
X>n
If X > n, stored value
= X
Else Input Error
Default
Some default values are constants, others are set by Phaser after it has analysed
the input data.
Single Use
Keywords marked single use are only applicable once. If more than one keyword
of this type is entered, the last value input will be used by Phaser.
Multiple Use
Keywords marked multiple use are meaningful when entered multiple times. The order
may or may not be important (see description of keyword for details in each case).
5. Python Scripting
As an alternative to keyword input, Phaser can be called directly from a python
scipt, because the core Phaser modes have been made available to python using
the Boost library. The syntax of the calls mirrors the keyworded input and is
easy to use. Users will need to have Phaser installed from source to have access
to the python scripting. For more information, please email
cimr-phaser@lists.cam.ac.uk.
beta_blip_ano.py:
Python script for anisotropy correction of beta-blip
from phaser import *
i = InputMR_DAT()
i.setHKLI("beta_blip.mtz")
i.addLABI("Fobs","Sigma")
i.Analyse()
o = Output()
r = ResultMR_DAT()
r = runMR_DAT(i,o)
hall = r.getHall()
cell = r.getUnitCell()
fobs = r.getFobs()
sigf = r.getSigFobs()
hkls = r.getMiller()
print r.logfile()
i = InputMR_ANO()
i.setSPAC_HALL(hall)
i.setCELL(cell[0],cell[1],cell[2],cell[3],cell[4],cell[5])
i.setREFL(hkls,fobs,sigf)
i.setROOT("beta_blip_ano")
i.Analyse()
r = ResultANO()
r = runMR_ANO(i,o)
print r.logfile()
6. References
- Read, R.J. (2001). Pushing
the boundaries of molecular replacement with maximum likelihood. Acta
Cryst. D57, 1373-1382
- Storoni, L.C, McCoy, A.J. & Read, R.J. (2004).
Likelihood
enhanced fast rotation functions. Acta Cryst D59, 1145-1153
