
| | Register New User | Download | Documentation | Tutorials | FAQ | Top Tips | Bugs | Publications | Developers |
Phaser is crystallographic software for phasing macromolecular crystal structures with maximum likelihood techniques. It is available through the Phenix and CCP4 software suites, and directly from the authors.
Most people will not need to read this documentation to solve their structure! To solve a structure by molecular replacement go to Automated Molecular Replacement and copy and edit the command script. Similarly, for SAD phasing, go to Automated Experimental Phasing and copy and edit the command script.
Other good sources of information are found in Frequently Asked Questions and Top Tips
This is the documentation for Phaser–2.0. There are some changes between this version and previous versions so input scripts may need editing.
Phaser runs in different modes, which perform Phaser's different functionalities. Modes can either be basic modes or modes that combine the functionality of basic modes.
The mode of operation is controlled with the MODE keyword
| Functionality | Mode | Description |
| Anisotropy Correction | ANO | Corrects data for anisotropic diffraction, and makes the intensity distribution isotropic |
| Cell Content Analysis | CCA | Calculates the expected number of molecular assemblies of given molecular weight in the unit cell using the Matthews coefficient |
| Normal Mode Analysis | NMA | Perturbs a structure in rms deviation steps along combinations of normal modes |
| Automated Molecular Replacement | MR_AUTO | Combines anisotropy correction, cell content analysis, fast rotation and translation functions, and refinement and phasing to automatically solve a structure by molecular replacement |
| Fast Rotation Function | MR_FRF | Anisotropy correction and likelihood-enhanced fast rotation function calculated with Fast Fourier Transform |
| Fast Translation Function | MR_FTF | Anisotropy correction and likelihood-enhanced fast translation function calculated with Fast Fourier Transform |
| Brute Rotation Function | MR_BRF | Anisotropy correction and full likelihood rotation function calculated in a brute-force search of angles |
| Brute Translation Function | MR_BTF | Anisotropy correction and full likelihood translation function calculated in a brute-force search of positions |
| Packing | MR_PAK | Tests molecular replacement solutions to see whether they pack into the unit cell without overlap |
| Log-Likelihood Gain | MR_LLG | Anisotropy correction and re-scoring of molecular replacement solutions with the full likelihood target function |
| Refinement and Phasing | MR_RNP | Anisotropy correction and optimization of the orientation and position of molecular replacement models with the full likelihood target function |
| Automated Experimental Phasing | EP_AUTO | Combines anisotropy correction, cell content analysis, and SAD phasing to automatically solve a structure by experimental phasing |
| SAD Phasing | EP_SAD | Refines atoms using the SAD likelihood function, and completes the structure with log-Likelihood gradient maps |
The example scripts all refer to the tutorial test cases. The pdb, sequence and mtz files required to run the tutorials are distributed with Phaser.
MODE ANO corrects the experimental data for anisotropy. Data (amplitude and associated sigma) are corrected for anisotropy and output to FILEROOT.mtz with column label set to the input column label with the addition of _ISO.
Example command script to correct BETA-BLIP data for anisotropy
beta_blip_ano.comCompulsory Keywords
Optional Keywords
MODE CCA determines the composition of the crystals using the "new" Matthews coefficients of Kantardjieff & Rupp (2003) "Matthews coefficient probabilities: Improved estimates for unit cell contents of proteins, DNA and protein-nucleic acid complex crystals". Protein Science 12:1865-1871. The molecular weight of ONE complex or assembly to be packed into the asymmetric unit is given with the COMPosition keyword, and the possible Z values (number of copies of the complex or assembly) that will fit in the asymmetric unit and the relative frequency of their corresponding VM values is reported. RESOlution should be set to the maximum resolution that has been observed for the crystal.
Example script for cell content analysis for BETA-BLIP
beta_cca.comCompulsory Keywords
Optional Keywords
MODE NMA writes out pdb files that have been perturbed along normal modes, in a procedure similar to that described by Suhre & Sanejouand (Acta Cryst. D60, 796-799, 2004). Each run of the program writes out a matrix FILEROOT.mat that contains the eigenvectors and eigenvalues of the atomic Hessian, and can be read into subsequent runs of the same job, to speed up the analysis.
Do normal mode analysis only, write out eigenfile but not coordinates
beta_nma.comWrite out pdb files perturbed in 0.5 Ångstrom rms intervals in "forward" (positive dq values) along modes 7 and 10 (and combinations of 7 and 10)
beta_nma_pdb.comCompulsory Keywords
Optional Keywords
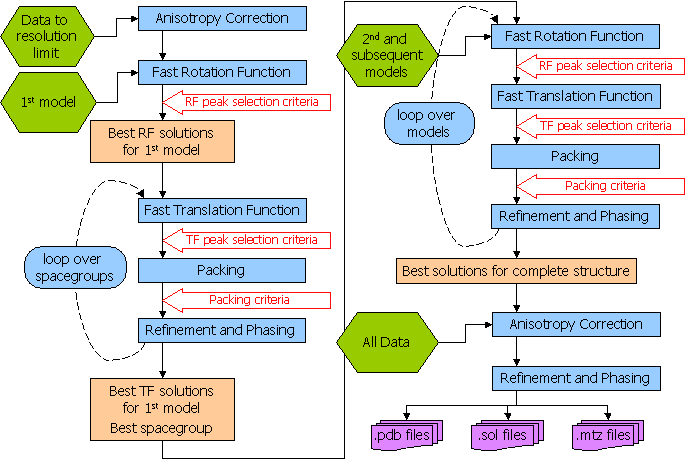
Phaser should be able to solve most structures with the Automated Molecular Replacement mode, and this is the first mode that you should try. Give Phaser your data (How to Define Data) and your models (How to Define Models), tell Phaser what to search for (use SEARch keyword), and a list of possible spacegroups (in the same pointgroup - use the SGALternative keyword). The flow diagram for the automated molecular replacement mode is shown below. If this doesn't work (see "Has Phaser Solved It?"), you can try selecting peaks of lower significance in the rotation function in case the real orientation was not within the selection criteria. By default peaks above 75% of the top peak are selected (see "How to Select Peaks"). See "What to do in difficult cases" for more hints and tips. If the automated molecular replacement mode doesn't work even with non-default input you need to run the modes of Phaser separately. The possibilities are endless - you can even try exhaustive searches (translations of all orientations) if you want - but experience has shown that most structures that can be solved by Phaser can be solved by relatively simple strategies.
MODE MR_AUTO combines the anisotropy correction, likelihood enhanced fast rotation function, likelihood enhanced fast translation function, packing and refinement modes for multiple search models and a set of possible spacegroups to automatically solve a structure by molecular replacement. Top solutions are output to the files FILEROOT.sol, FILEROOT.#.mtz and FILEROOT.#.pdb (where "#" refers to the sorted solution number, 1 being the best, and only 1 is output by default). Many structures can be solved by running an automated molecular replacement search with defaults, giving the ensembles that you expect to be easiest to find first.
Example command script for finding BETA and BLIP. This is the minimum input, using all defaults (except the ROOT filename).
beta_blip_auto.comExample command script for finding BETA and BLIP. The spacegroup recorded on the mtz file is P3221 but the other hand is also a possibility. Both search orders (BETA first, BLIP second and BLIP first, BETA second) are tried, using the PERMutations ON keyword. We would not normally recommend using the PERMutations ON keyword for this case, as it is obvious that the larger molecule should be easier to find first. To speed up the calculation only the top peak after the translation function is taken into refinement.
beta_blip_auto_sg.comCompulsory Keywords
Optional Keywords
Ideally, only the number of solutions you are expecting should be found. However if the signal-to-noise of your search is low, there will be noise peaks in the final selection also.
A highly compact summary of the history of a solution is given in the annotation of a solution in the .sol file. This is a good place to start your analysis of the output. The annotation gives the Z-score of the solution at each rotation and translation function, the number of clashes in the packing, and the refined LLG. You should see the TFZ (the translation function Z-score) is high at least for the final components of the solution, and that the LLG (log-likelihood gain) increases as each component of the solution is added. For example, in the case of beta-blip the annotation for the single solution output in the .sol file shows these features
SOLU SET RFZ=11.0 TFZ=22.6 PAK=0 LLG=434 RFZ=6.2 TFZ=28.9 PAK=0 LLG=986 LLG=986
SOLU 6DIM ENSE beta EULER 200.920 41.240 183.776 FRAC -0.49641 -0.15752 -0.28125
SOLU 6DIM ENSE blip EULER 43.873 80.949 117.141 FRAC -0.12290 0.29306 -0.09193
You should always at least glance through the summary of the logfile. One thing to look for, in particular, is whether any translation solutions with a high Z-score have been rejected by the packing step, especially with a small number of clashes. Such a solution may be correct, and the clashes may arise only because of differences in small surface loops. If this happens, repeat the run allowing a suitable number of clashes with the PACK keyword. Note that, unless there is specific evidence in the logfile that a high TF-function Z-score solution is being rejected with a few clashes, it is much better to edit the model to remove the loops than to increase the number of allowed clashes. Packing criteria are a very powerful constraint on the translation function, and increasing the number of allowed clashes beyond a few (e.g. 1-5) will increase the search time enormously without the possibility of generating any correct solutions that would not have otherwise been found.
For a rotation function, the correct solution may be in the list with a Z-score under 4, and will not be found until a translation function is performed and picks out the correct solution.
For a translation function the correct solution will generally have a Z-score (number of standard deviations above the mean value) over 5 and be well separated from the rest of the solutions. Of course, there will always be exceptions! Note, in particular, that in the presence of translational NCS, pairs of similarly-oriented molecules separated by the correct translation vector will give large Z-scores, even if they are incorrect, because they explain the systematic variation in intensities caused by the translational NCS.
| TF Z-score |
Have I solved it? |
| less than 5 |
no |
| 5 - 6 |
unlikely |
| 6 - 7 |
possibly |
| 7 - 8 |
probably |
| more than 8 |
definitely |
The relative orientations of the domains may be different in your crystal than in the model. If that may be the case, break the model into separate PDB files containing rigid-body units, enter these as separate ensembles, and search for them separately. If you find a convincing solution for one domain, but fail to find a solution for the next domain, you can take advantage of the knowledge that its orientation is likely to be similar to that of the first domain. The ROTAte AROUnd option of the brute rotation search can be used to restrict the search to orientations within, say, 30 degrees of that of the known domain. Allow for close approach of the domains by increasing the allowed clashes with the PACK keyword by, say, 1 for each domain break that you introduce.
Alternatively, you could try generating a series of models perturbed by normal modes, with the NMAPdb keyword. One of these may duplicate the hinge motion and provide a good single model.
Signal-to-noise is reduced by coordinate errors or incompleteness of the model. Since the rotation search has lower signal to begin with than the translation search, it is usually more severely affected. For this reason, it can be very useful to use the subsequent translation search as a way to choose among many (say 1000) orientations. Try increasing the number of clustered orientations in an AUTO job using the keyword FINAL, e.g. FINAL ROT SELEct PERCent 65. If that fails, try turning off the clustering feature in the save step (FINAL ROT CLUSter OFF), because the correct orientation may sit on the shoulder of a peak in the rotation function.
As shown convincingly by Schwarzenbacher et al. (Schwarzenbacher, Godzik, Grzechnik & Jaroszewski, Acta Cryst. D60, 1229-1236, 2004), judicious editing can make a significant difference in the quality of a distant model. In a number of tests with their data on models below 30% sequence identity, we have found that Phaser works best with a "mixed model" (non-identical sidechains longer than Ser replaced by Ser). In agreement with their results, the best models are generally derived using more sophisticated alignment protocols, such as their FFAS protocol.
The automated mode of Phaser is fast when Phaser finds a high Z-score solution to your problem. When Phaser cannot find a solution with a significant Z-score, it "thrashes", meaning it maintains a list of 100-1000's of low Z-score potential solutions and tries to improve them. This can lead to exceptionally long Phaser runs (over a week of CPU time). Such runs are possible because the highly automated script allows many consecutive MR jobs to be run without you having to manually set 100-1000's of jobs running and keep track of the results. "Thrashing" generally does not produce a solution: solutions generally appear relatively quickly or not at all. It is more useful to go back and analyse your models and your data to see where improvements can be made. Your system manager will appreciate you terminating these jobs.
It is also not a good idea to effectively remove the packing test by setting the allowed number of clashes with the packing test to a high number (e.g PACK 1000). Unless there is specific evidence in the logfile that a high TF-function Z-score solution is being rejected with a few clashes, it is much better to edit the model to remove the loops than to increase the number of allowed clashes. Packing criteria are a very powerful constraint on the translation function, and increasing the number of allowed clashes beyond a few (e.g. 1-5) will increase the search time enormously without the possibility of generating any correct solutions that would not have otherwise been found.
Phaser is has powerful input, output and scripting facilities that allow a large number of possibilities for altering default behaviour and forcing Phaser to do what you think it should. However, you will need to read the information in the manual below to take advantage of these facilities!
You need to tell Phaser the name of the mtz file containing your data and the columns in the mtz file to be used using the HKLIn and LABIn keywords. Additional keywords (BINS CELL OUTLier RESOlution SPACegroup) define how the data are used.
Phaser must be given the models that it will use for molecular replacement. A molecular replacement model is constructed in one of two ways - either by making an ensemble from a set of aligned structures, entered as pdb files, or by entering a model from a map, entered as structure factors in an mtz file. Each ensemble is treated as a separate type of rigid body to be placed in the molecular replacement solution. An ensemble should only be defined once, even if there are several copies of the molecule in the asymmetric unit.
Fundamental to the way in which Phaser uses MR models (either from coordinates or maps) is to estimate
how the accuracy of the model falls off as a function of resolution, represented by the Sigma(A) curve.
To generate the Sigma(A) curve, Phaser needs to know the
RMS coordinate error expected for the model and the fraction of the scattering power in the asymmetric unit
that this model contributes.
If fp is the fraction scattering and RMS is the rms coordinate
error, then
Sigma(A) = SQRT{fp*[1-fsol*exp(-Bsol*(sin(theta)/lambda)2)]}
* exp{-(8 Pi2/3)*RMS2*(sin(theta)/lambda)2}
where fsol(default=0.95) and Bsol(default=300Å2) account for the effects
of disordered solvent on the completeness of the model at low resolution.
Molecular replacement models are defined with the ENSEmble keyword and the COMPosition keyword. The ENSEmble keyword gives (amongst other things) the RMS deviation for the Sigma(A) curve. The COMPosition keyword is used to deduce the fraction of the scattering power in the asymmetric unit that each ensemble contributes. The composition of the asymmetric unit is defined either by entering the molecular weights or sequences of the components in the asymmetric unit, and giving the number of copies of each. Expert users can also enter the fraction of the scattering of each component directly, although the composition must still be entered for the absolute scale calculation.
The RMS deviation is determined directly from RMS or indirectly from IDENtity in the ENSEmble keyword using the formula RMS = max(0.8,0.4*exp(1.87*(1.0-ID))) where ID is the fraction identity. The RMS deviation estimated from ID may be an underestimate of the true value if there is a slight conformational change between the model and target structures. To find a solution in these cases it may be necessary to increase the RMS from the default value generated from the ID, by say 0.5 Ångstroms. The table below can be used as a guide as to the default RMS value corresponding to ID.
| Sequence ID |
RMS deviation |
| 100% |
0.80Å |
| 64% |
0.80Å |
| 63% |
0.799Å |
| 50% |
1.02Å |
| 40% |
1.23Å |
| 30% |
1.48Å |
| 20% |
1.78Å |
| --> limit 0% |
2.60Å |
If you construct a model by homology modelling, remember that the RMS error you expect is essentially the error you expect from the template structure (if not worse!). So specify the sequence identity of the template, not of the homology model.
When using density as a model, it is necessary to specify both the extent (x,y,z limits) of the cut-out region of density, and the centre of this region. With coordinates, Phaser can work this out by itself. This information is needed, for instance, to decide how large rotational steps can be in the rotation search and to carry out the molecular transform interpolation correctly. In the case of electron density, the RMS value does not have the same physical meaning that it has when the model is specified by atomic coordinates, but it is used to judge how the accuracy of the calculated structure factors drops off with resolution. A suitable value for RMS can be obtained, in the case of density from an experimentally-phased map, by choosing a value that makes the SigmaA curve fall off with resolution similar to the mean figures-of-merit. In the case of density from an EM image reconstruction, the RMS value should make the SigmaA curve fall off similar to a Fourier correlation curve used to judge the resolution of the EM image.
For detailed information, including tutorial with example scripts, see Using density as a model
The composition is the total amount of protein and nucleic acid that you have in the asymmetric unit not the fraction of the asymmetric unit that you are searching for.
The composition is calculated from the molecular weight of the protein and nucleic acid assuming the protein and nucleic acid have the average distribution of amino acids and bases. If your protein or nucleic acid has an unusual amino acid or base distribution the composition should be entered by sequence. You can mix compositions entered by molecular weight with those entered by sequence.
The composition is calculated from the amino acid sequence of the protein and the base sequence of the nucleic acid in fasta format. You can mix compositions entered by molecular weight with those entered by sequence. Individual atoms can be added to the composition with the COMPOSITION ATOM keyword. This allows the explicit addition of heavy atoms in the structure e.g. Fe atoms.
The fraction scattering of each ensemble can be entered directly. The fraction scattering of each ensemble is normally automatically worked out from the average scattering from each ensemble (calculated from the pdb files if entered as coordinates, or from the protein and nucleic acid molecular weights if entered as a map) divided by the total scattering given by the composition, but entering the fraction scattering directly overrides this calculation. This option is for use when the pdb files of the models in the ensemble are unusual e.g. consist only of C-alpha atoms, or only of hydrogen atoms (as in the CLOUDS method for NMR).
Phaser writes out files ending in ".sol" and ".rlist" that contain the solution information from the job. The root of the files is given by the ROOT keyword. By default, the root filename is PHASER. These files can be read back into subsequent runs of Phaser to build up solutions containing more than one molecule in the asymmetric unit.
"PHASER.sol" files are generated by all modes (rotation function modes with VERBOSE output), and contain the current idea of potential molecular replacement solutions.
"PHASER.rlist" files are generated by the rotation function modes, and are for performing translation functions. (They are also produced by degenerate (2D) translation functions, for performing a translation function to find the third dimension)
To include the files you should use the preprocessor command @
For simple MR cases you don't really need to know how to define molecular replacement solutions. However, for difficult cases you might need to edit the files "PHASER.sol" and "PHASER.rlist" files manually
At different stages of molecular replacement, an Ensemble will be oriented but not positioned (after the rotation search), or oriented and positioned (after the translation search), or, rarely, oriented and the position in 2 of 3 dimensions known. These three states correspond to solutions defined by the keywords SOLUtion 3DIM, SOLUtion 6DIM, and SOLUtion 5DIM. Each Ensemble in the asymmetric unit has its own SOLUtion keyword. Solutions of the type 3DIM are given by the rotation function, solutions of the type 6DIM are given by the translation function, and solutions of the type 5DIM are given by the degenerate translation function. Examples are:
When more than one (potential) molecular replacement solution is present, the solutions are separated with the SOLUTION SET keywords. For example, if the rotation function and translation function for mol1 were very clear, then there will only be one type of 6DIM solution for mol1. If the rotation and translation functions for mol2 were then not clear, there will be a series of possible 6DIM solutions for mol2.
If you have the coordinates of a partial solution with the pdb coordinates of the known structure in the correct orientation and position, then you can force Phaser to use these coordinates by manually creating a .sol file of the following form and including it in the Phaser command script with the @filename preprocessor command (or including it directly in the script)
These files define a rotation function list. The peak list is given with a series of SOLUtion TRIAl keywords.
If a partial solution is already known, then the information for the currently "known" parts of the asymmetric unit is given in the form used for the PHASER.sol file, followed by the list of trial orientations for which a translation function is to be performed.
If a degenerate translation function is performed, then a SOLUtion TRIAl line is produced with the degenerate translation information present, ready for performing the translation function on the third dimension.
The output of Phaser can be controlled with the following optional keywords. The ROOT keyword is not compulsory (the default root filename is "PHASER"), but should always be given, so that your jobs have separate and meaningful output filenames.
Optional Keywords
Where HKLOut ON is given as an optional keyword, Phaser produces an mtz file with "SigmaA" type weighted Fourier map coefficients for producing electron density maps for rebuilding.
| MTZ Column Labels | Description | |
| FWT | PHWT | Amplitude and phase for 2m|Fobs|-D|Fcalc| exp(i alpha-calc) map |
| DELFWT | PHDELWT | Amplitude and phase for m|Fobs|-D|Fcalc| exp(i alpha-calc) map |
| FOM | m, analogous to the "Sim" weight, to estimate the reliability of alpha-calc | |
The selection of peaks for the fast rotation and fast translation function with rescoring of the top peaks with the full likelihood target (default RESCORE ON), is done in three steps, controlled by the keyword FINAL
For automated molecular replacement, specifying [ROT|TRA] after the keyword determines which of the rotation or translation function the selection criteria apply to. If neither is specified, the selection criteria apply to both.
| Keyword | Applies |
| FINAL [ROT|TRA] STEP 1 | Controls the selection of peaks from the fast search that will be rescored with the full likelihood target |
| FINAL [ROT|TRA] STEP 2 | Controls the selection of peaks from the rescoring to be combined with other searches (e.g translation functions of different rotations, or rotation functions with different fixed components present) |
| FINAL [ROT|TRA] STEP 3 | Controls the selection of peaks from the merged list for final output |
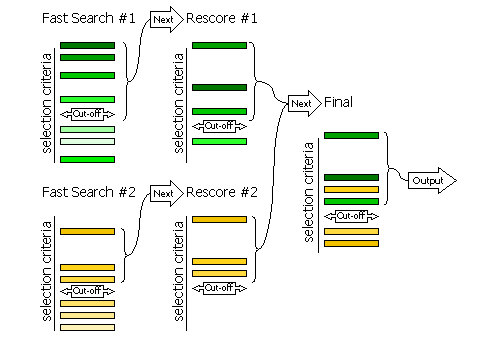
If RESCORE OFF is requested (no rescoring of the fast search peaks is performed), or if the brute rotation or translation searches are carried out, then there are only two stages to selection: selection of peaks from the individual searches, and selection of peaks from the combined list of solutions. Selection of peaks at each stage is controlled, respectively, by the keywords FINAL [ROT|TRA] STEP 2 and FINAL [ROT|TRA] STEP 3.
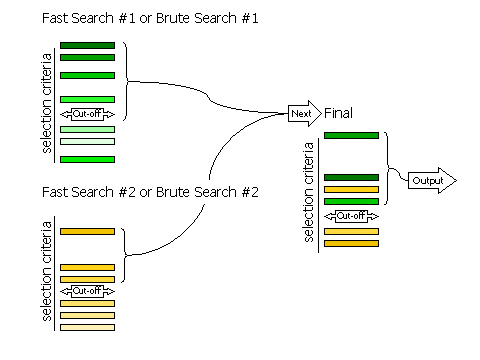
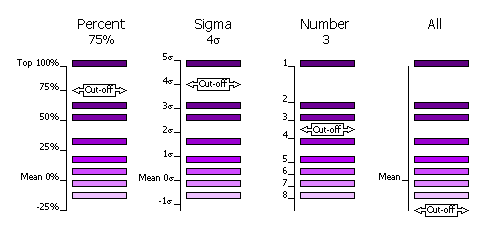
The selection of peaks saved for output in the rotation and translation functions can be done in four different ways.
| Sub-Keyword | Description | Use |
| FINAL [ROT|TRA] STEP [1|2|3] SELEct PERCent <CUTOFF> | Percentage of the top peak, where the value of the top peak is defined as 100% and the value of the mean is defined as 0%. | Default, cutoff=75%. This criteria has the advantange that at least one peak (the top peak) always survives the selection. If the top solution is clear, then only the one solution will be output, but if the distribution of peaks is rather flat, then many peaks will be output for testing in the next part of the MR procedure (e.g. many peaks selected from the rotation function for testing with a translation function). |
| FINAL [ROT|TRA] STEP [1|2|3] SELEct SIGma <CUTOFF> | Number of standard deviations (sigmas) over the mean (the Z-score) | Absolute significance test. Not all searches will produce output if the cutoff value is too high (e.g. 5 sigma). |
| FINAL [ROT|TRA] STEP [1|2|3] SELEct NUMber <CUTOFF> | Number of top peaks to select | If the distribution is very flat then it might be better to select a fixed large number (e.g. 1000) of top rotation peaks for testing in the translation function. |
| FINAL [ROT|TRA] STEP [1|2|3] SELEct ALL | None: all peaks are selected | Enables full 6 dimensional searches, where all the solutions from the rotation function are output for testing in the translation function. This should never be necessary; it would be much faster and probably just as likely to work if the top 1000 peaks were used in this way. |
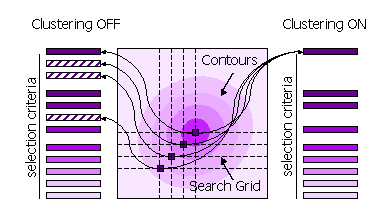
Peaks can also be clustered or not clustered prior to selection in steps 1 and 2.
| Sub-Keyword | Description | Use |
| FINAL [ROT|TRA] STEP [1|2] CLUSTER OFF | All high peaks on the search grid are selected | Default for STEP 1, because the position of the maximum may be different when the fast score and the full likelihood function are used. |
| FINAL [ROT|TRA] STEP [1|2] CLUSTER ON | Points on the search grid with higher neighboring points are removed from the selection | Default for STEP 2. |
MODE MR_FRF combines the anisotropy correction and likelihood-enhanced fast rotation function (2), optionally rescored with the full rotation likelihood function (1), to find the orientation of a model in molecular replacement. Top rotation solutions are output to the file FILEROOT.rlist for input to a translation function. Top rotation solutions are also output to the file FILEROOT.sol.
Example command script for fast rotation function to find the orientation of BETA.
beta_frf.comExample command script for fast rotation function to find the orientation of BLIP knowing the position and orientation of BETA, with the position and orientation of BETA input from the command line.
blip_frf_with_beta.comExample command script for fast rotation function to find the orientation of BLIP knowing only the orientation of BETA, with the orientation of BETA input using the output solution file from the beta_frf.com job above.
blip_frf_with_beta_rot.comCompulsory Keywords
Optional Keywords
MODE MR_BRF combines the anisotropy correction and brute force likelihood rotation function (1) to find the orientation of a model in molecular replacement. Top rotation solutions are output to the file FILEROOT.rlist for input to a translation function. Top rotation solutions are also output to the file FILEROOT.sol.
Example command script for brute rotation function to find the orientation of BETA
beta_brf.comExample command script for brute rotation function to find the optimal orientation of BETA in a restricted search range and on a fine grid around the position from the fast rotation search.
beta_brf_around.comCompulsory Keywords
Optional Keywords
MODE MR_FTF combines the anisotropy correction and likelihood-enhanced fast translation function (3), optionally rescored by the full likelihood translation function (1), to find the position of a previously oriented model in molecular replacement. Top translation solutions are output to the file FILEROOT.sol.
Example command script for finding the position of BETA after the rotation function has been run and the results output to the file beta_frf.rlist
beta_ftf.comExample command script for finding the position of BLIP after the rotation function has been run and the results output to the file blip_frf_with_beta.rlist, which has the SOLUtion 6DIM keyword input for BETA and the SOLUtion TRIAL keyword input for the orientations to try for BLIP with the translation function.
blip_ftf_with_beta.comCompulsory Keywords
Optional Keywords
MODE MR_BTF combines the anisotropy correction and brute force likelihood translation function (1) to find the position of a previously oriented model in molecular replacement. Top translation solutions are output to the file FILEROOT.sol.
Example command script for brute Translation function to find the position of BETA after the rotation function has been run
beta_btf.comExample command script for brute Translation function to find the position of BETA degenerate in X after the rotation function has been run
beta_btf_degen_x.comCompulsory Keywords
Optional Keywords
MODE MR_RNP combines the anisotropy correction and refinement against the likelihood function (1) to optimize full or partial molecular replacement solutions and phase the data. At the end of refinement, the list of solutions is checked for duplicates, which are pruned. Refined solutions are output to the file FILEROOT.sol.
Example command script to refine a set of solutions
beta_blip_rnp.comCompulsory Keywords
Optional Keywords
MODE MR_LLG combines the anisotropy correction and the likelihood function (1) to calculate the log-likelihood gain for full or partial molecular replacement solutions. Solutions are output to the file FILEROOT.sol.
Example command script to rescore the solutions using a different resolution range of data and a different spacegroup
beta_blip_llg.comCompulsory Keywords
Optional Keywords
MODE MR_PAK determines whether molecular replacement solutions pack in the unit cell. Solutions that pack are output to the file FILEROOT.sol.
Example command script for determining whether a set of molecular replacement solutions pack in the unit cell
beta_blip_pak.comCompulsory Keywords
Optional Keywords
Phaser performs SAD phasing in two modes. In the Automated Experimental Phasing mode, Phaser corrects for anisotropy, puts the data on absolute scale, does a cell content analysis, refines heavy atom sites to optimize phasing, and completes the model from log-likelihood gradient maps. Alternatively, the SAD Phasing mode can be used, which only refines heavy atom sites to optimize phasing, and completes the model from log-likelihood gradient maps. For this mode, the data should be pre-corrected for anisotropy and put on an absolute scale. This mode should only be used as part of automation pipelines, where the correct preparation of the data can be guaranteed and it saves cpu time.
MODE EP_AUTO combines the anisotropy correction, cell content analysis, and SAD Phasing modes to automatically solve a structure by experimental phasing. The final solution is output to the files FILEROOT.sol, FILEROOT.mtz and FILEROOT.pdb. Many structures can be solved by running an automated experimental phasing job with defaults.
Do SAD phasing of insulin. This is the minimum input, using all defaults (except the ROOT filename) insulin_auto.comCompulsory Keywords
Optional Keywords
You need to tell Phaser the name of mtz file containing your data and the columns in the mtz file to be used. For SAD phasing, a single CRYSTAL and DATASET with anomalous data (F(+), SIGF(+), F(-) and SIGF(-)) must be given. The columns must have the correct CCP4 column type: 'G' for F(+) and F(-) and 'L' for SIGF(+) and SIGF(-). If the columns on your mtz file have somehow acquired the incorrect column type, you should change the column type with an mtz editing programme (e.g. sftools).
CRYStal insulin DATAset sad &Compulsory Keywords
Optional Keywords
Atom sites are defined with the ATOM keyword. Atoms sites may be entered one at a time specifying fractional or orthogonal coordinates, occupancy and B-factor, or from a PDB file, or from a mlphare-style HA file. The crystal to which the atoms correspond must be specified in the input.
The output of Phaser can be controlled with the following keywords:
Optional Keywords
MODE EP_SAD phases SAD data and completes the structure from log-likelihood gradient maps. The final solution is output to the files FILEROOT.sol, FILEROOT.mtz and FILEROOT.pdb .
Do SAD phasing of insulin. This is the minimum input, using all defaults (except the ROOT filename)
insulin_sad.comCompulsory Keywords
Optional Keywords
Phaser can be controlled using keyword input. Not all keywords are relevant for all modes of operation (the list of relevant keywords for each mode is given with each mode above). Some keywords are only for single use, others have meaning when used more than once. The input values of many parameters are constrained to physically meaningful values. All non-compulsory parameters have defaults.
Preprocessor commands may be used in the keyword input to incorporate files, add comments or allow line continuation.
Most keywords only refer to a single parameters, and if used multiple times, the parameter will take last value input. Some keywords are meaningful when entered multiple times. The order may or may not be important.
Phaser can generate XML output from keyword input. XML output should be used in preference to grepping logfiles when incorporating Phaser into automation pipelines. Note that Phaser's python scripting ability is a more powerful way of calling Phaser for automation pipelines.
We would like to hear from developers who wish to incorporate Phaser into automation scripts using the XML functionality cimr-phaser@lists.cam.ac.uk
Phaser outputs an XML file when called with the command line argument -xml followed by the filename for output.
phaser -xml <filename>
If no filename is given, Phaser exits immediately and writes an XML file with filename PHASER.XML describing the error (type="FILE OPENING", message="No XML filename").
All XML output is wrapped between "phaser" tags with the version number for the Phaser executable and the operating system on which the output was produced as attributes.
Names of files are output using the "file" tags with attributes "type" specifying reflections ("HKL") or coordinates ("XYZ") and attribute "format". Currently only "MTZ" and "PDB" formats are output.
Other XML tags are a combination of those suggested by the SPINE consortium and Phaser specific tags. There is no schema. Please refer to the examples.
Successful Phaser execution (not necessarily structure solution!) is reported as
Failure during execution is reported as
More information about the type of error is given with
Allowed values for ERROR_NAME are given in the table below, and specify the type of error. The ERROR_MESSAGE gives more information as to the specific cause of the error.
| ERROR_NAME | Failure due to … |
| SYNTAX | Syntax error in keyword input |
| INPUT | Input error (e.g. invalid value for an input parameter) |
| FILE OPENING | Unable to open file (given in ERROR_MESSAGE) for reading or writing. |
| OUT OF MEMORY | Memory exhaustion |
| FATAL RUNTIME | Fatal runtime error (e.g. bug in Phaser) |
| UNHANDLED | Other unhandled fatal error (e.g. bug in libraries) |
| UNKNOWN | Other error (e.g. bug in compiler) |
Use the keyword MUTE ON to prevent the writing of the logfile to standard output. Only the XML file and other results files (mtz,pdb) will be produced by Phaser.
Below are example XML output files produced by running the most popular modes of Phaser: anisotropy correction, automated molecular replacement, cell content analysis, normal mode analayis, and SAD phasing.
Output XML file for anisotropy correction of BETA-BLIP
<phaser version="2.0" ostype="linux">Output XML file for Cell Content Analysis of BETA-BLIP
<phaser version="2.0" ostype="linux">Output XML file for Normal Mode Analysis of beta.pdb (modes 7 and 10, displacement forward only)
<phaser version="2.0" ostype="linux">Output XML file for Automated Molecular Replacement of BETA-BLIP
<phaser version="2.0" ostype="linux">Output XML file for SAD phasing of insulin
<phaser version="2.0" ostype="linux">As an alternative to keyword input, Phaser can be called directly from a python script. This is the way Phaser is called in Phenix and we encourage developers of other automation pipelines to use the python scripting too. In order to call Phaser in python you will need to have Phaser installed from source.
We would like to hear from developers who wish to incorporate Phaser into automation scripts using the python scripting functionality cimr-phaser@lists.cam.ac.uk
| Functionality | Input-Object | Run-Job | Results-Object |
| Anisotropy Correction | i = InputANO() | r = runANO(i) | ResultANO() |
| Cell Content Analysis | i = InputCCA() | r = runCCA(i) | ResultCCA() |
| Normal Mode Analysis | i = InputNMA() | r = runNMA(i) | ResultNMA() |
| Automated MR | i = InputMR_AUTO() | r = runMR_AUTO(i) | ResultMR() |
| Fast Rotation Function | i = InputMR_FRF() | r = runMR_FRF(i) | ResultMR_RF() |
| Brute Rotation Function | i = InputMR_BRF() | r = runMR_BRF(i) | ResultMR_RF() |
| Fast Translation Function | i = InputMR_FTF() | r = runMR_FTF(i) | ResultMR_TF() |
| Brute Translation Function | i = InputMR_BTF() | r = runMR_BTF(i) | ResultMR_TF() |
| Refinement and Phasing | i = InputMR_RNP() | r = runMR_RNP(i) | ResultMR() |
| Log-Likelihood Gain | i = InputMR_LLG() | r = runMR_LLG(i) | ResultMR() |
| Packing | i = InputMR_PAK() | r = runMR_PAK(i) | ResultMR() |
| Automated Experimental Phasing | i = InputEP_AUTO() | r = runEP_AUTO(i) | ResultEP() |
| SAD Experimental Phasing | i = InputEP_SAD() | r = runEP_SAD(i) | ResultEP() |
The major difference between running Phaser though the keyword interface and running Phaser though the python scripting is that the data reading and Phaser functionality are separated. For the Phaser "run-job" functions, the reflection data (for Miller indices, Fobs and SigmaFobs) are simply arrays, the spacegroup is given as a Hall string, and the unitcell is given as an array of 6 numbers. This is an important feature of the Phaser python scripting as it means that the Phaser "run-job" functions are not tied to mtz file input, but the data can be read in python from any file format, and then the data passed to Phaser.
For the convenience of developers and users, the python scripting comes with data-reading jiffies to read data from mtz files. (These are the same mtz reading jiffies that are used internally by Phaser when calling Phaser from keyword input.)
| Functionality | Input-Object | Run-Job | Result-Object |
| Read Data for MR | i = InputMR_DAT() | r = runMR_DAT(i) | ResultMR_DAT() |
| Read Data for EP | i = InputEP_DAT() | r = runEP_DAT(i) | ResultEP_DAT() |
Note that setting the spacegroup by name or number does not specify the setting. It is best to set the spacegroup via the Hall symbol, which is unique to the full definition of the spacegroup.
| Input Objects | Python Set Function | |
| ROOT filename | i.setROOT(filename) | |
| MUTE [ON|OFF] | i.setMUTE(True|False) | |
| TITLe title | i.setTITL(title) | |
| VERBose [ON|OFF] | i.setVERB(True|False) | |
| VERBose [ON|OFF] EXTRA | i.setVERB_EXTRA(True|False) | |
| * | SPACegroup name | i.setSPAC_NAME(name) |
| * | SPACegroup number | i.setSPAC_NUM(number) |
| * | SPACegroup Hall | i.setSPAC_HALL(hall) |
| * | CELL a b c alpha beta gamm | i.setCELL(a,b,c,alpha,beta,gamma) |
| * | Cell set from array of 6 numbers | i.setCELL([a,b,c,alpha,beta,gamma]) |
| * except InputNMA | ||
Data are extracted from the "result-objects" with get-functions. The get-functions are mostly specific to the type of "result-object" (described in sections below), but some are common to all "result-objects" (described in table below).
Ralf Grosse-Kunstleve's scitbx::af::shared<double> array type is heavily used for passing of arrays into the Phaser "input-objects" and extracting arrays from the Phaser "result-objects". This is a reference counted array type that can be used directly in python and in C++. It is part of the Phaser installation, when Phaser is installed from source. The scitbx (SCIentific ToolBoX) is part of the cctbx (Computational Crystallography ToolBoX) which is hosted by sourceforge
| Results Objects | Python Get Function | |
| Exit status "success" | r.Success() | |
| Exit status "failure" | r.Failure() | |
| Type of Error (see error table). SYNTAX errors are not thrown in python as they are generated by keyword input | r.ErrorName() | |
| Message associated with error | r.ErrorMessage() | |
| Text of Summary | r.summary() | |
| Text of Logfile | r.logfile() | |
| Text of Verbose Logfile | r.verbose() | |
| * | SpaceGroup Hall Symbol | r.getSpaceGroupHall() |
| * | SpaceGroup Name (Hermann Mauguin, edited for CCP4 compatibility in R3 H3 R32 H32) | r.getSpaceGroupName() |
| * | SpaceGroup Number | r.getSpaceGroupNumber() |
| * | Number of symmetry operators | r.getSpaceGroupNSYMM() |
| * | Number of primative symmetry operators | r.getSpaceGroupNSYMP() |
| * | Symmetry operator #s, Rotation matrix element i,j (range 0-2) | r.getSpaceGroupR(s,i,j) |
| * | Symmetry operator #s, Translation vector element i (range 0-2) | r.getSpaceGroupT(s,i) |
| * | Unit Cell (array of 6 numbers) | r.getUnitCell() |
| * except ResultNMA | ||
Exit status is indicated by Success() and Failure() functions of the "result-objects". Success indicates successful execution of Phaser, not that it has solved the structure! For molecular replacement jobs, the foundSolutions() function indicates that Phaser has found one or more potential solutions, the numSolutions() function returns how many solutions were found and the uniqueSolution() function returns True if only one solution was found. More detailed error information in the case of Failure is given by ErrorName() and ErrorMessage().
Advanced Information: All errors are thrown and caught internally by the "run-jobs", and so do not generate "Runtime Errors" in the python script. In particular "INPUT" errors are not thrown by the set- or add-functions of the "input-objects", but are stored in the "input-object" and passed to the "result-object" once the "run-job" is called. Results objects are derived from std::exception, and so can be thrown. Function what() returns ErrorName() (not the ErrorMessage()).
Writing of the logfile to standard output can be silenced with the i.setMUTE(True) function. The logfile or summary text can then be printed to standard output with the print r.logfile() or print r.summary() functions.
Advanced Information: Setting i.setMUTE(True) prevents real time viewing of the progress of a Phaser job. This may present an inconvenience for users. If you want to view the logfile information but not have it go to standard output, Logfile text can be redirected to a python string using an alternative call to the "run-job" function that includes passing an "output-object" (which controls the Phaser logging methods) on which the output stream has been set to a python string. This feature of Phaser was developed thanks to Ralf Grosse-Kunstleve.
beta_blip_logfile.pyBelow are the detailed instructions for running the most popular modes of Phaser: anisotropy correction, automated molecular replacement, cell content analysis, normal mode analysis, and SAD phasing. Note that not all the functionality is available through the python interface, particularly the "expert" functionality available through the keyword interface, marked with an asterix in the keyword list.
Input Object Type: InputMR_DAT
| Keyword Input | Python Set/Add Function |
| HKLIn filename | i.setHKLI(filename) |
| LABIn F=Fobs SIGF=Sigma | i.setLABI(Fobs,Sigma) |
| RESOlution lim | i.setHIRES(lim) |
| RESOlution lim1 lim2 | i.setRESO(lim1,lim2 |
| SORT [ON|OFF] | i.setSORT(True|False) |
Result Object Type: ResultMR_DAT
| Result | Python Get Function |
| Miller Indices (array) | r.getMiller() |
| F values (array) | r.getF() |
| SIGF values (array) | r.getSIGF() |
Example script for reading data from MTZ file beta_blip.mtz
Note that by default reflections are sorted into resolution
order upon reading, to achieve a performance gain in the molecular replacement routines. If reflections are not being read from
an MTZ file with this script, reflections should be pre-sorted into resolution order to achieve the same performance gain. Sorting is turned off with the setSORT(False) function.
Input Object Type: InputANO
| Keyword Input | Python Set/Add Function |
| Read from MTZ file | i.setREFL(HKL,F,SIGF) |
| HKLIn filename MTZ file to which output is appended Data are not read from this file |
i.setHKLI(filename) |
| LABIn F=Fobs SIGF=Sigma Column label in output MTZ file Fobs_ISO and Sigma_ISO Defaults F_ISO and SIGF_ISO |
i.setREFL_ID(Fobs,Sigma) |
| HKLOut [ON|OFF] | i.setHKLO(True|False) |
| SORT [ON|OFF] | i.setSORT(True|False) |
Result Object Type: ResultANO
| Result | Python Get Function |
| Miller Indices (array) | r.getMiller() |
| F values (array) | r.getF() |
| SIGF values (array) | r.getSIGF() |
| Corrected F (array) | r.getCorrectedF() |
| Corrected SIGF (array) | r.getCorrectedSIGF() |
| Correction Factor | r.getCorrection() |
| Apply scale and correction factors to array | new_array = r.getScaledCorrected(array) |
| Factor to put data on absolute scale | r.WilsonK() |
| Wilson B factor | r.WilsonB() |
| Measure of anisotropy | r.getAnisoDeltaB() |
| Eigenvalues of anisotropy | r.getEigenBs() |
| Eigenvectors and Eigenvalues or anisotropy | r.getEigenSystem() |
| Name of output MTZ file | r.getMtzFile() |
| Output MTZ file corrected F label | r.getLaboutF() |
| Output MTZ file corrected SIGF label | r.getLaboutSIGF() |
| Name of output XML file | r.getXmlFile() |
Example script script for anisotropy correction of BETA-BLIP data
beta_blip_ano.py
Input Object Type: InputCCA
| Keyword Input | Python Set/Add Function |
| COMPosition PROTein MW mw NUM num | i.addCOMP_PROT_MW_NUM(mw,num) |
| COMPosition PROTein SEQ file NUM num | i.addCOMP_PROT_FASTA_NUM(file,num) where sequence in a fasta file i.addCOMP_PROT_SEQ_NUM(sequence,num) where sequence as a string, one letter code |
| COMPosition NUCLeic MW mw NUM num | i.addCOMP_NUCL_MW_NUM(mw,num) |
| COMPosition NUCLeic SEQ seq NUM num | i.addCOMP_NUCL_FASTA_NUM(file,num) where sequence in a fasta file i.addCOMP_NUCL_SEQ_NUM(sequence,num) where sequence as a string, one letter code |
| COMPosition ATOM atomtype NUM num | i.addCOMP_ATOM_NUM(atomtype,num) |
| COMPosition ENSEmble ens FRAC frac | i.addCOMP_ENSE_FRAC(ens,frac) |
Result Object Type: ResultCCA
| Result | Python Get Function |
| Molecular weight of the assembly used for VM calculations | r.getAssemblyVM() |
| Number of multiples of the assembly within allowed VM range | r.getNum() |
| Array of the multiples (Z) of the assembly within allowed VM range | r.getZ() |
| Array of the values of VM corresponding to the multiples (Z) of the assembly | r.getVM() |
| Array of the probabilities of VM corresponding to the multiples (Z) of the assembly | r.getProb() |
| Most probable multiple (Z) of the assembly | r.getBestZ() |
| VM of the most probable multiple (Z) of the assembly | r.getBestVM() |
| Probability of the most probable multiple (Z) of the assembly | r.getBestProb() |
| XML file name | r.getXmlFile() |
| Optimal VM for spacegroup, unitcell and resolution | r.getOptimalVM() |
| Optimal MW for spacegroup, unitcell and resolution | r.getOptimalMW() |
Example script for cell content analysis of BETA-BLIP
beta_blip_cca.py
Input Object Type: InputNMA
| Keyword Input | Python Set/Add Function |
| ENSEmble … | As for InputMR_AUTO |
| NMAPdb MODE m1 MODE m2 … | i.setNMAP_MODES([m1,m2…]) |
| NMAPdb RMS step | i.setNMAP_RMS(step) |
| NMAPdb MAXRms distance | i.setNMAP_MAXRMS(distance) |
| NMAPdb BACKward | i.setNMAP_BACKWARD() |
| NMAPdb FORWard | i.setNMAP_FORWARD() |
| NMAPdb TOFRo | i.setNMAP_TO_AND_FRO() |
| NMAPdb COMBination nmax | i.setNMAP_COMBINATION(nmax) |
| NMAPdb CLASh distance | i.setNMAP_CLASH(distance) |
| NMAPdb STREtch distance | i.setNMAP_STRETCH(distance) |
| NMAPdb DQ dq1 DQ dq2 … | i.setNMAP_DQ([dq1,dq2…]) |
| NMAMethod RTB | i.setNMAM_RTB() |
| NMAMethod CA | i.setNMAM_CA() |
| NMAMethod ALL | i.setNMAM_ALL() |
| NMAMethod NRESidues nres | i.setNMAM_NRES(nres) |
| NMAMethod MAXBlocks maxblocks | i.setNMAM_MAXBLOCKS(maxblocks) |
| NMAMethod RADIus radius | i.setNMAM_RADIUS(radius) |
| NMAMethod FORCe force | i.setNMAM_FORCE(force) |
| SCRIpt [ON|OFF] | i.setSCRI(True|False) |
| XYZOut [ON|OFF] | i.setXYZO(True|False) |
Result Object Type: ResultNMA
| Result | Python Get Function |
| Number of total perturbations along combinations of normal modes | r.getNum() |
| Array of all pdb files | r.getPdbFiles() |
| Name of pdb file for perturbation #i | r.getPdbFile(i) |
| Array of normal modes contributing to perturbation #i | r.getModes(i) |
| Array of displacements along modes contributing to perturbation #i | r.getDisplacements(i) |
| Script output file | r.getSolFile() |
| Xml output file | r.getXmlFile() |
Example script for normal mode analysis of BETA-BLIP. Note that the spacegroup and unitcell are not required, and so the MTZ file does not need to be read to extract these parameters.
beta_nma.py
Input Object Type: InputMR_AUTO
| Keyword Input | Python Set/Add Function |
| Read from MTZ file | i.setREFL(HKL,F,SIGF) |
| SGALternative num | i.addSGAL_NUM(num) |
| SGALternative name | i.addSGAL_NAME(name) |
| SGALternative HAND | i.addSGAL_HAND(True|False) |
| SGALternative ALL | i.addSGAL_ALL(True|False) |
| ENSEmble ens PDB pdbfile ID id | i.addENSE_PDB_ID(ens,pdbfile,id) |
| ENSEmble ens PDB pdbfile RMS rms | i.addENSE_PDB_RMS(ens,pdbfile,rms) |
| COMPosition … | As for InputCCA |
| SEARch ENSEmble ens NUM num | i.addSEAR_ENSE_NUM(ens,num) |
| SEARch ENSEmble ens1 {OR ENSEmble …} NUM num | i.addSEAR_ENSE_OR_ENSE_NUM([ens1,…],num) |
| HKLOut [ON|OFF] | i.setHKLO(True|False) |
| SCRIpt [ON|OFF] | i.setSCRI(True|False) |
| XYZOut [ON|OFF] | i.setXYZO(True|False) |
Result Object Type: ResultMR
| Result | Python Get Function |
| Solutions were found (boolean) | r.foundSolutions() |
| Number of Solutions that were found (int) | r.numSolutions() |
| Only one solution found (boolean) | r.uniqueSolution() |
| LLG values for all solutions in decreasing order | r.getValues() |
| Script output file | r.getSolFile() |
| Xml output file | r.getXmlFile() |
| PDB files corresponding to solutions in decreasing LLG order | r.getPdbFiles() |
| MTZ files corresponding to solutions in decreasing LLG order | r.getMtzFiles() |
| LLG of top solution | r.getTopValue() |
| PDB file of top solution | r.getTopPdbFile() |
| MTZ file of top solution | r.getTopMtzFile() |
| List of details of solutions (rotation, translation) in decreasing LLG order in "mr_solution" type |
r.getDotSol() |
Example script for automated structure solution of BETA-BLIP
beta_blip_auto.py
Input Object Type: InputEP_DAT
| Keyword Input | Python Set/Add Function |
| HKLIn filename | i.setHKLI(filename) |
| LABIn F=Fobs SIGF=Sigma | i.setLABI(Fobs,Sigma) |
| RESOlution lim | i.setHIRES(lim) |
| RESOlution lim1 lim2 | i.setRESO(lim1,lim2) |
| CRYStal xtal DATAset wave LABIn F = F SIGF = SIGF | i.addCRYS_NORM_LABI(xtal,wave,F,SIGF) |
| CRYStal xtal DATAset wave LABIn F+ = Fp SIG+ = SIGp F- = Fn SIG- = SIGn | i.addCRYS_ANOM_LABI(xtal,wave,Fp,SIGp,Fn,SIGn) |
| CRYStal xtal DATAset wave RESOlution lim1 lim2 | i.setCRYS_RESO(xtal,wave,lim1,lim2) |
Result Object Type: ResultEP_DAT
| Result | Python Get Function |
| Miller Indices (array) | r.getMiller() |
| Non-anomalous F values for crystal "xtal" and dataset "wave" (array) | r.getF(xtal,wave) |
| Non-anomalous SIGF values for crystal "xtal" and dataset "wave" (array) | r.getSIGF(xtal,wave) |
| Boolean flags for F (and SIGF) present for crystal "xtal" and dataset "wave" (array) | r.getP(xtal,wave) |
| Anomalous F+ values for crystal "xtal" and dataset "wave" (array) | r.getFpos(xtal,wave) |
| Anomalous SIGF+ values for crystal "xtal" and dataset "wave" (array) | r.getSIGFpos(xtal,wave) |
| Boolean flags for F+ (and SIGF+) present for crystal "xtal" and dataset "wave" (array) | r.getPpos(xtal,wave) |
| Anomalous F- values for crystal "xtal" and dataset "wave" (array) | r.getFneg(xtal,wave) |
| Anomalous SIGF- values for crystal "xtal" and dataset "wave" (array) | r.getSIGFneg(xtal,wave) |
| Boolean flags for F- (and SIGF-) present for crystal "xtal" and dataset "wave" (array) | r.getPneg(xtal,wave) |
Below is a python script for reading SAD data from MTZ file S-insulin.mtz
Input Object Type: InputEP_AUTO
| Keyword Input | Python Set/Add Function |
| HKLIn filename
MTZ to which output data are appended Data are not read from this file |
i.setHKLI(filename) |
| Input Data from array | i.setCRYS_MILLER(miller) |
| Input Non-anomalous data for xtal/wave from array | i.addCRYS_NORM_DATA(xtal,wave,F,SIGF,P) |
| Input Anomalous data for xtal/wave from array | i.addCRYS_ANOM_DATA(xtal,wave,Fp,SIGp,Pp,Fn,SIGn,Pn) |
| CRYS xtal DATA wave SCAT CUKA | i.setCRYS_SCAT_CUKA(xtal,wave) |
| CRYS xtal DATA wave SCAT WAVE lambda | i.setCRYS_SCAT_WAVE(xtal,wave,lambda) |
| CRYS xtal DATA wave SCAT MEAS FP=fp FDP=fdp | i.setCRYS_SCAT_MEAS(xtal,wave,fp,fdp) |
| CRYS xtal DATA wave SCAT ATOM type FP=fp FDP=fdp | i.addCRYS_SCAT_ELEM(xtal,wave,type,fp,fdp) |
| CRYS xtal DATA wave SCAT FIXP FIXDP | i.addCRYS_SCAT_FIX(xtal,wave,True,True) |
| LLGC CRYS xtal COMPLETE [ON|OFF] | i.setLLGC_CRYS_COMPLETE(xtal,True|False) |
| LLGC CRYS xtal SCATTERING ELEMENT type | i.addLLGC_CRYS_SCAT_ELEMENT(xtal,type) |
| LLGC CRYS xtal CLASH distance | i.setLLGC_CRYS_CLASH(xtal,distance) |
| LLGC CRYS xtal SIGMA cutoff | i.setLLGC_CRYS_SIGMA(xtal,cutoff) |
| ATOM CRYS xtal ELEM type FRAC x y z OCC occ | i.addATOM(xtal,type,x,y,z,occ) |
| ATOM BFAC WILSON | i.setATOM_BFACTOR_WILSON() |
| ATOM BFAC VALUE bfactor | i.setATOM_BFACTOR_VALUE(bfactor) |
| ATOM CRYS xtal PDB pdbfile | i.setATOM_PDB(xtal,pdbfile) |
| ATOM CRYS xtal HA hafile | i.setATOM_HA(xtal,hafile) |
| HAND [ON|OFF] | i.setHAND(True|False) |
| COMPosition … | As for InputCCA |
Result Object Type: ResultEP
| Result | Python Get Function |
| Log-likelihood of refined solution | r.getLogLikelihood() |
| Miller Indices (array) | r.getMiller() |
| Boolean array flagging reflections included in electron denisty | r.getSelected() |
| Figures of merit for phased dataset (array) | r.getFOM() |
| Amplitudes for weighted electrion density of phased dataset (array) | r.getFWT() |
| Phases for weighted electrion density of phased dataset (array) | r.getPHWT() |
| Phases for electrion density of phased dataset (array) | r.getPHIB() |
| Amplitudes for log-likelihood gradient map | r.getFLLG() |
| Phases for log-likelihood gradient map | r.getPHLLG() |
| Atoms included in final solution for crystal xtal | r.getAtoms(xtalid) |
| Atoms rejected from final solution for crystal xtal | r.getRejectedAtoms(xtalid) |
| f' for atomtype "type" in crystal "xtald" dataset "wave" | r.getFp(xtalid,wave,type) |
| f" for atomtype "type" in crystal "xtald" dataset "wave" | r.getFdp(xtalid,wave,type) |
| Name of output MTZ file | r.getMtzFile() |
| Name of output PDB file | r.getPdbFile() |
| Name of output SOL file | r.getSolFile() |
| Name of output XML file | r.getXmlFile() |
There are also functions for extracting phasing statistics
| Result | Python Get Function |
| Overall low resolution limit | r.stats_lores() |
| Overall high resolution limit | r.stats_hires() |
| Overall figure of merit for all reflections | r.stats_fom() |
| Overall figure of merit for acentrics | r.stats_acentric_fom() |
| Overall figure of merit for centrics | r.stats_centric_fom() |
| Overall figure of merit for singleton | r.stats_singleton_fom() |
| Overall number of reflections | r.stats_num() |
| Overall number of acentric reflections | r.stats_acentric_num() |
| Overall number of centric reflections | r.stats_centric_num() |
| Overall number of singleton reflections | r.stats_singleton_num() |
| Number of resolution bins for statistics | r.stats_numbins() |
| Binwise low resolution limit | r.stats_by_bin_lores(bin_number) |
| Binwise high resolution limit | r.stats_by_bin_hires(bin_number) |
| Binwise middle of resolution range | r.stats_by_bin_midres(bin_number) |
| Binwise figure of merit for all reflections | r.stats_by_bin_fom(bin_number) |
| Binwise figure of merit for acentrics | r.stats_by_bin_acentric_fom(bin_number) |
| Binwise figure of merit for centrics | r.stats_by_bin_centric_fom(bin_number) |
| Binwise figure of merit for singleton | r.stats_by_bin_singleton_fom(bin_number) |
| Binwise number of reflections | r.stats_by_bin_num(bin_number) |
| Binwise number of acentric reflections | r.stats_by_bin_acentric_num(bin_number) |
| Binwise number of centric reflections | r.stats_by_bin_centric_num(bin_number) |
| Binwise number of singleton reflections | r.stats_by_bin_singleton_num(bin_number) |
Example script for SAD phasing for insulin
insulin_sad.py